
Systems biology of the structural proteome
Genome-scale models of metabolism (GEM) represent biochemical, genetic and genomic (BiGG) knowledge bases that can be utilised in a wide variety of theoretical and practical computational studies. These may range from the discovery of unidentified metabolic reactions, to the exploration of host/pathogen interactions. Recently, these models have been extended via the incorporation of additional biological information, such as protein structural information, thus giving birth to genome-scale models with protein structures (GEM-PRO).
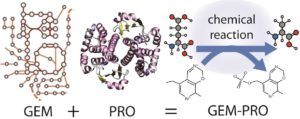
This article generates and applies the GEM-PRO methodology on two distinct organisms: Escherichia coli and Thermotoga maritima. It is a multi-scale attempt that creates multiple links between genes (and their products), biochemical reactions and phenotypic functions, that are further enhanced by molecular-level information about individual proteins. Essentially, GEM-PRO lies at the intersection of systems biology and structural biology and offers insight into the physical embodiment of an organism’s genotype.
To aid in the understanding and further application of this methodology specific tutorials showing how protein-related information can be linked to genome-scale models and can be accessed in a public GitHub repository (https://github.com/SBRG/GEMPro/tree/master/GEMPro_recon/).
Feedback control in planarian stem cell systems
Planarian flatworms have astonishing regenerative mechanisms in response to their environmental conditions and for that property they have been systematically studied for the past 100+ years. However, currently it is possible to trace their stem cell activity from the gene-level all the way to the organism-level, providing a unique opportunity for a ‘systems biology’ approach.
This article presents a nonlinear dynamical model of the flatworm’s stem cell system incorporating feedback control. It draws conclusions on the dynamics of the size, signaling systems and rates of mortality for the aforementioned animal and builds the foundations for a full conceptual framework for planarian cellular dynamics. Based on this model, the scientific community may begin to understand the mechanisms of cell migration during injury, the characterization of homeostatic levels of differentiated cells, as well as the associated stochastic effects.
Comparing Alzheimer’s and Parkinson’s diseases networks using graph communities structure
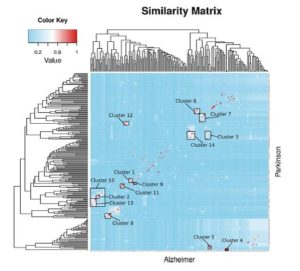
The rapid advancement of high throughput technologies, along with the vast amounts of available ‘omics’ data, cultivate the ideal environment for a system-level investigation of pathologies. In this article, Calderone et al. showcase a state-of-the-art algorithm, termed ‘InfoMap’, which is able to exploit network community structures and is built upon network theory fundamentals.
It is applied to two age-related neurodegenerative diseases, Alzheimer’s Disease and Parkinson’s Disease, in order to identify similarities and differences in their respective network proteins and pathways. Significant insights are gained for both known and unknown processes, but particularly with regards to information about mitochondrial dysfunction and metabolism. The remarkable fact about this methodological approach is that it can be applied to the comparison of any pair of biological networks.
Where next for the reproducibility agenda in computational biology?
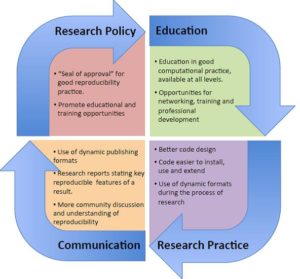
The term ‘reproducibility’ is a gray aspect for the computational biology community. Unfortunately, it is often (wrongly) open to interpretation and can have a spectrum of definitions. The authors of this study claim that the most basic form of reproducibility is replicability; the notion that “do other people get exactly the same results when doing exactly the same thing?” (i.e. method replicability). However, true reproducibility requires not only the method but also the phenomenon itself to be reproducible. Finally, when reproducing a software result the ultimate aim it to build on it, so researchers need to also consider the extensibility of their computational method.
The authors present case studies that provide insight on these three aspects. They argue that well-designed software needs to facilitate these aspects in order to ensure the value, quality and assurance of the presented computational work. Finally, specific checklists and recommendation lists are provided to guide researchers, developers and even the community towards improving and ensuring reproducibility in computational biology.
Integrating mutation and gene expression cross-sectional data to infer cancer progression
Reproducibility is certainly an issue when it comes to cancer research and that’s due to the heterogeneity of the disease. In biomarker discovery certain patterns of ‘omics’ data in one study may fail to be validated in another. In clinical terms, that can be attributed to a number of factors, ranging from the employed molecular methodology and the existing cancer data sets, all the way to the poorly modular patient diagnosis and the complex dynamics of cancer progression.
Fleck et al propose a novel systematic methodology to infer temporal sequence by integrating gene mutation with gene expression data. They are able to identify a set of mutation events that may eventually lead to changes in the gene expression. Furthermore, this model is specifically tested on both simulated and real breast cancer data from The Cancer Genome Atlas. Overall, by identifying the groups of genes that change during cancer progression this model may provide superior insight to the aforementioned heterogeneity of cancer, further address clinical questions or even improve the current therapeutic strategies.
Comments