
deep molecular generative models … enable large amounts of molecules to be generated from a small set of known ones
One of cheminformatics’ main goals is to design novel molecules by exploring the whole chemical space. This virtual chemical space is comprised of all possible molecules, and by some estimated to be around 10^60 molecules.
Until recently, chemical space exploration was done by generating molecules using genetic algorithms or through joining together smaller molecules to form bigger ones.
A different, still ongoing, approach is the generated database (GDB) project, which aims to enumerate all molecules up to a certain number of non-hydrogen atoms. To date, the drug-like chemical space up to 11, 13 and 17 atoms has been enumerated and yielded databases with 25 million, 1 billion and 165 billion molecules respectively.
These huge databases can then be searched to find novel molecules of interest. As the chemical space grows exponentially with the number of atoms, exploring larger regions of it becomes an ever-increasingly difficult endeavor.
As in many disciplines of science, deep learning and artificial intelligence (AI) have opened new perspectives in cheminformatics. For example, we can train neural networks with data from heterogeneous sources to predict the toxicity of molecules, or we can design algorithms that obtain all the steps required to synthesize any molecule.
But one of the most impressive advances to date has been deep molecular generative models, which enable large amounts of molecules to be generated from a small set of known ones.
In particular, recurrent neural networks (RNNs) trained with molecules represented in a text format called SMILES have proven very successful in exploring the chemical space.
They work similarly to the autocomplete function of cellphone keyboards and build molecules character-by-character taking into account the already generated partial molecule (Figure 1). As models learn how to create molecules just from the training data, generated molecules tend to show the same properties as those in the training set.

Therefore, the chemical space around the training set can be explored by repeatedly generating (also called sampling) molecules from an RNN.
One of the drawbacks of molecular generative models is that they are sampled with replacement: when the model is sampled more than one time, repeated molecules may appear. This can yield a situation in which the model seems to generate a diverse set of molecules, but it is generating the same set repeatedly.
Alternatively, a model can generate a huge amount of molecules completely unrelated to the training set, implying that the model does not learn from the training set molecules. In our research, we developed a benchmarking method that is able to detect both situations.
To be able to do that, we train models on a subset of the previously mentioned GDB-13 molecular database, we sample the model 2 billion times and we calculate how many unique generated molecules are part of GDB-13 and how many outside of it. The models are then ranked by how much of the whole database they can generate when learning from a small sample of it.
We used this method to benchmark several molecular generative models with two ways of representing molecules: canonical and randomized SMILES. This text-based representation is built by numbering the atoms in the molecule and then traversing it, adding a “C” when it’s a carbon, an “O” for an oxygen, etc. (Figure 2).
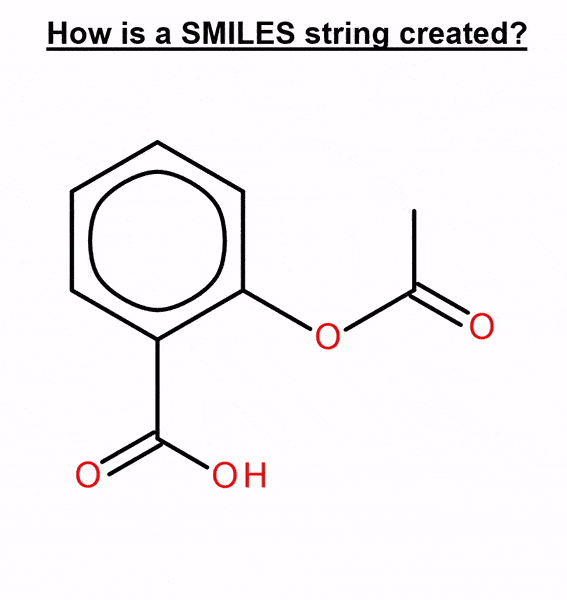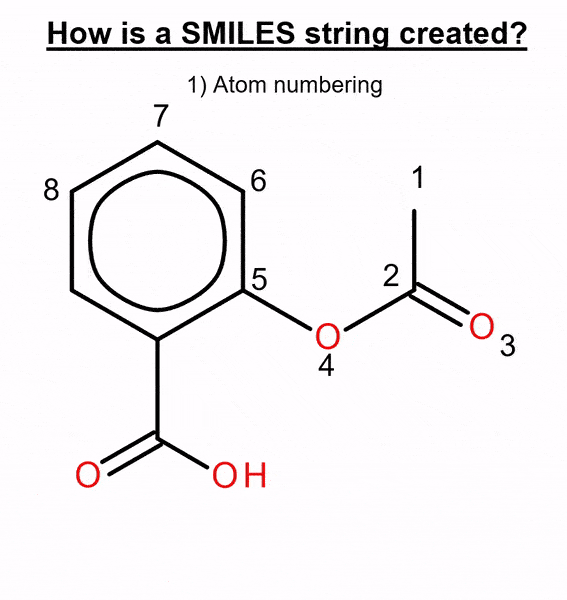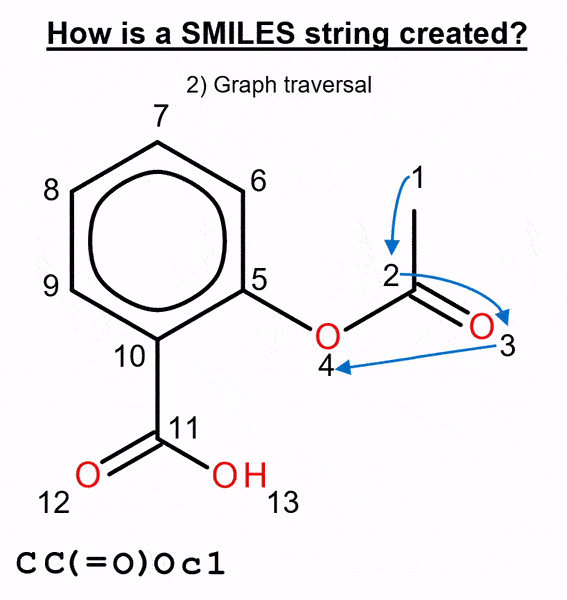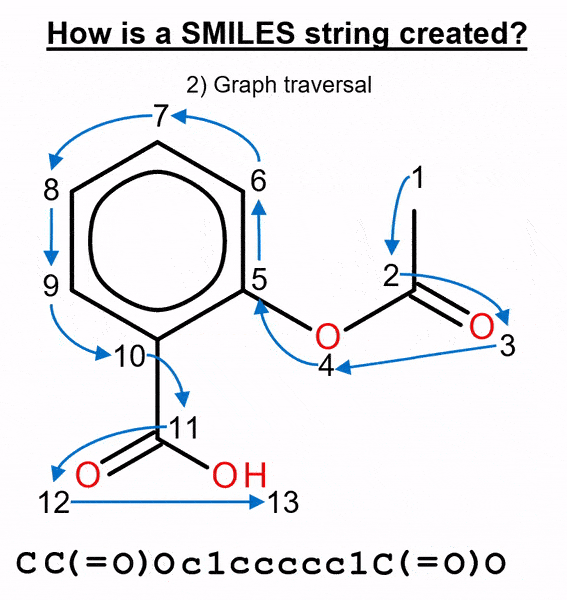
By default, most chemistry software calculates a unique atom numbering, called canonical ordering, and the canonical SMILES is generated from it. When this atom numbering is randomized, different SMILES strings that represent the same molecule can be obtained, thus obtaining randomized SMILES.
During training, models using canonical SMILES always use the same representation for each molecule, whereas those using randomized SMILES keep changing it. This allows the models to see the same molecule in different angles, learning different information every time.
When comparing models trained with a subset of one million GDB-13 molecules (0.1% of the database) with canonical and randomized SMILES, results show that canonical SMILES models are able to generate up to 70% of GDB-13, whereas randomized SMILES models work substantially better, obtaining up to 83% of the whole database.
Smaller training sets were also used, and when a randomized SMILES model was trained with 1000 molecules (0.0001% of GDB-13), 34% of the whole database was obtained compared to only 14% generated with a canonical SMILES model.
Given the improvement obtained with randomized SMILES, one last experiment was carried out on models trained with the ChEMBL database. This database holds a 1.5 million selection of molecules obtained from literature that represent a sample the whole known drug-like chemical space.
Results showed that models trained with randomized SMILES generated circa 1.3 billion unique molecules from a 2 billion sample, which was nearly double the amount of molecules than with canonical SMILES.
Furthermore, we also showed that any molecule generated by the canonical SMILES model could be generated by the randomized SMILES model but not vice versa.
In conclusion, generative models trained with a small molecular sample are capable of probabilistically holding a large slice of the chemical space. Then, trained models can be exploited, either by extensive sampling and filtering, or by using techniques such as reinforcement learning.
Having architectures that can convey as much information as possible from small training sets to the generated molecules is extremely important in drug discovery and randomized SMILES are an improvement that helps on that direction.
These developed methods are currently used in many drug discovery projects in AstraZeneca, and we hope to report back to the scientific community the results applying them to in-house projects in due course.

Comments