The emerging realization that cells modify the three dimensional arrangement of DNA in order to regulate the genome is changing the way that scientists think about how and when genes are expressed. A new study in Genome Biology goes so far as to show that information about the shape of the genome is sufficient on its own to accurately classify cells according to leukemia subtype.
Traditional genomics studies have examined the genome as a linear DNA sequence. According to this viewpoint, each gene is surrounded by adjacent sequence – in particular the region of the genome immediately upstream – that regulates its expression: when the gene is turned off and on, and to what degree.
It has long been known that more distant sequence in the genome can also regulate genes, but recent work has established a greater importance for this phenomenon, which is mediated by direct contacts made by looping and twisted DNA – requiring us to think about the genome in three dimensions.
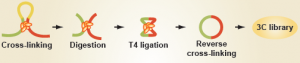
For many years, genomics studies had shied away from analyzing these three dimensional regulatory events for a very good reason: we had no easy way of knowing where distal contacts were occurring. However, the emergence of a new set of technologies, collectively known as 'chromatin conformation capture' (with variants known as '3C', '4C', '5C' and 'HiC'), has now enabled researchers to map long-range gene regulation to – at times – quite startling effect.
Predicting leukemia subtype

As discussed in a Biome Research Synopsis, Josée Dostie and colleagues now report in Genome Biology that the shape of the genome, as determined by 5C, can be as predictive – if not more so – than gene expression in classifying leukemia subtype by MLL fusion and fusion partner.
The FTO theory loses weight

Pity the poor PhD researcher who has been assigned a project on the FTO gene and weight loss. After multiple genome-wide association studies pointed the finger at this gene as a candidate influencer of obesity risk, scientists set out to determine the mechanistic basis for FTO making people fat – but such a mechanism turned out to be frustratingly elusive.
Along came Marcelo Nóbrega and colleagues, armed with 4C technology and an interest in long-range gene regulation. They found that the obesity phenotype had precisely nothing to do with the protein encoded by the FTO gene, but instead was due to contact made by an intron in FTO with a gene located several megabases away, IRX3.
This 3D arrangement of the genome appears to be important in ensuring that IRX3 is appropriately regulated. And it is the protein encoded by IRX3, rather than FTO, that explains the observed association with obesity.
Characterizing long-range regulatory regions
Is there a way to identify regulatory regions of the genome that act at long range without mapping the 3D shape of DNA? Mathematical modelers have shown the predictive power of combining DNA sequence information with data on transcription factor binding – although the effect of sequence on DNA looping seems to be (at least partly) indirect, through changes in nucleosome organization. This leads us to the epigenome: recent research articles in Epigenetics & Chromatin and Genome Biology complemented chromatin conformation capture techniques with methylomics to characterize DNA methylation at distal regulatory sites.
With more data, machine-learning computational approaches may be able to identify diagnostic features, such as DNA methylation and nucleosome distribution, that are predictive of long-range regulatory function.
Interestingly, Asaf Hellman and colleagues used machine learning to identify the DNA methylation signatures of 'distal enhancers', which are expected to regulate genes through long-range contact, finding that dysregulation of DNA methylation at these sites is associated with cancer.
Dostie and colleagues also used machine learning in their study; how their chromatin conformation signatures fit together with the predictive power of DNA methylation and other epigenomic marks might be one future direction for further inquiry.
Transcription factories
An interesting theory, put forward by Genome Biology Editorial Board member Peter Fraser and others, to explain one way that genome shape might regulate gene expression is that of 'transcription factories'.
Transcription factories are proposed to be substructures in the nucleus that concentrate genes together from different parts of the genome – often even from different chromosomes. These substructures overflow with RNA polymerase enzymes in order to ensure intensive, coordinated gene expression.
Immunofluorescence experiments have been used to support the existence of transcription factories for select groups of genes, and it has even been suggested that some cancers are caused by aberrant fusions between genes making close contact at transcription factories – something to think about when considering the implications of Dostie and colleagues' leukemia study.
The dynamic 3D genome
A key requirement for regulatory mechanisms is of course the ability to react to changes in the cell and its surroundings.
Now that chromatin conformation capture technologies are becoming widely established, we can move forward from basic mapping of the 3D genome to examine how DNA becomes rearranged through the life cycle of the cell, as well as in response to various stimuli, and to consider what variation may exist in the 3D genome between cells – and why.
A recent Q&A on single-cell genomics in Biome highlighted the possibility of performing chromatin conformation capture experiments in single cells as one of the most exciting new directions in the field, while a recent Research Highlight in Genome Biology discussed the remarkable insights into genome shape rearrangement during the cell cycle yielded by HiC and 5C.
Examples such as these show how the impact of chromatin conformation capture technology looks set to be radical and disruptive, as we herald in the era of the dynamic 3D genome.
- tRFs and the Argonautes: gene silencing from antiquity - 2nd October 2014
- Keeping up with the Jobses: the role of technology in reproducible research - 26th September 2014
- How to disarm a superbug – a story told by forensic genomics - 23rd June 2014
One Comment