Chickens, like us, are vulnerable to a wide range of pests and pathogens. There are effective vaccines, drugs or disinfectants against some chicken diseases, but control of many others remains a distant prospect. Sequencing pathogen genomes can be a valuable step in developing new control measures. After more than ten years of work, we are pleased to report that genomes are now completed for the most important group of parasites that infect chickens. This new knowledge offers exciting prospects for better control of the globally devastating chicken gut disease, coccidiosis.

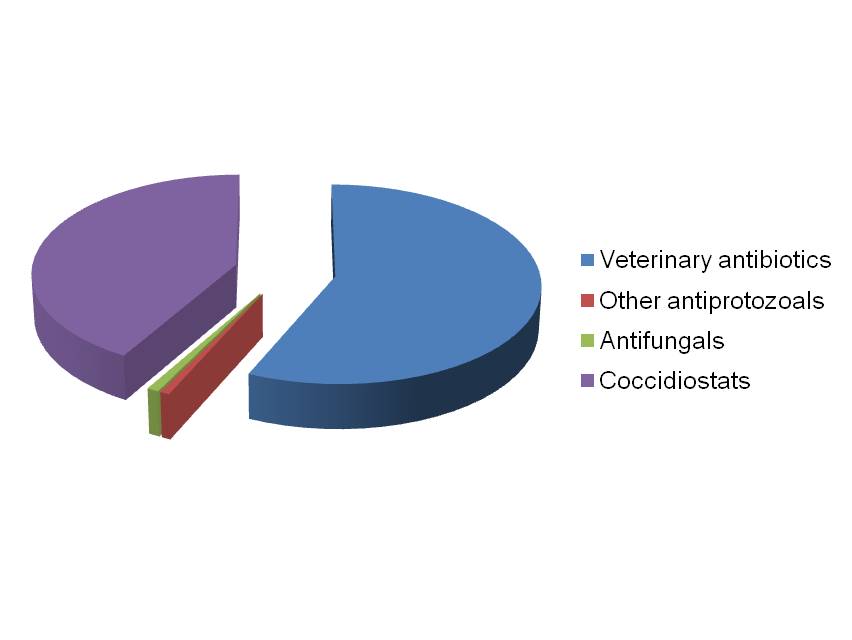
In a study between scientists at the Royal Veterinary College and Wellcome Trust Sanger Institute in the UK, the King Abdullah University of Science and Technology in Saudi Arabia, the University Kebangsaan in Malaysia and the University of São Paulo in Brazil, we sequenced and analysed genomes of all seven species of the Eimeria parasites which cause chicken coccidiosis. Chickens are the world’s most popular food animal. Global poultry production has tripled in the past 20 years and more than 60 billion chickens are now produced each year, providing 1.1 trillion eggs and 90 million tonnes of meat. Growth in the poultry sector is expected to continue for some years to come and the challenges associated with safe, welfare-friendly, sustainable meat and egg production are immense. A major concern is endemic infectious diseases that compromise bird health and welfare, and reduce productivity. Many of these ’production’ diseases pose serious threats to the global food supply chain and contribute directly to poverty in developing rural economies where smallholder livelihoods can be wiped out through disease. Eimeria are single-celled protozoa, closely related to the parasites that cause malaria, cryptosporidiosis and toxoplasmosis. Coccidiosis can affect all livestock but is most problematic in chickens, mainly because the parasites have a direct faecal/oral lifecycle and they spread rapidly when large numbers of hosts are housed in close quarters. If not controlled, coccidiosis causes severe enteric disease with very high rates of morbidity and mortality (Figure 1). Current control is based mainly on the use of anti-coccidial drugs, which account for more than 40% of the total antimicrobials usage in UK animals (Figure 2). Unsurprisingly, parasite genetic resistance to all classes of anti-coccidial drugs can occur and resistance is ubiquitous in all parts of the world, which means control by drugs is always sub-optimal and may be completely ineffective. Live parasite vaccines are also available, and whilst these work well they are relatively expensive, compared to drugs, and because they have to be produced in chickens there are severe constraints on the number of doses that can realistically be manufactured.
Seven Eimeria species can infect chickens, each colonising a preferred region of the intestine where they cause symptoms of differing severity. All seven species can cause disease but four (E. acervulina, E. maxima, E. necatrix and E. tenella) are of most importance because they occur frequently, replicate to high levels and are highly pathogenic. We generated high quality ‘complete’ genome sequences for these four species, and less comprehensive sequences for the remaining three species (E. brunetti, E. mitis, E. praecox), complimented in all cases by coding sequence annotations. The publication of these resources means the Eimeria genus has joined a league of sequenced apicomplexan protozoa which already includes species from the Cryptosporidium, Neospora, Plasmodium, Sarcocystis, Theileria and Toxoplasma genera. These resources are publically available through the web database EuPathDB , which offers a wide variety of bioinformatics tools to allow users to carry out in-depth analyses and studies of comparative genomics. It is the expectation that understanding the detailed comparative biology of these types of parasites will improve ‘rational’ development of new control measures by allowing reconstruction and interrogation of essential parasite metabolic pathways, identifying potential drug or vaccine targets and identification parasite genes under diversifying selection. We used a combination of Sanger and Illumina technologies to generate the genome sequences, with computer-based assembly supported by the physical technique of optical mapping. For E. tenella, the most thoroughly defined of all the Eimeria genomes we worked on, there are significantly more protein-coding genes predicted than in the closely related Toxoplasma gondii, despite E. tenella having a smaller nuclear genome.

A key finding from our analyses was the genus-wide conservation of repeat-rich/repeat-poor segments within Eimeria genomes, with repeats being more common in protein-coding sequences than reported for any other organisms in nature. The ubiquity of repeats in coding regions means that Eimeria parasites produce many proteins that contain widespread Homopolymeric Amino Acid Repeats (HAARs). A detailed gene ontology analysis did not identify any particular functional class associated with these HAARs; however “core” genes that are conserved amongst all known eukaryotes have fewer repeats than non-core genes. We also found that Eimeria genomes possess retrotransposon sequences, containing predominantly fragmented long terminal repeat (LTR) retrotransposons from the chromovirus group. This is the first time these have been found within an apicomplexan genome. All the Eimeria genomes contained families of genes that encode polymorphic, membrane-linked surface antigens (SAGs). These have been studied to some extent previously in E. tenella and shown to localise to the outermost membrane of the invading stages of parasites where they can interact directly with host cell surface molecules during the process of parasite attachment and host cell invasion. Comparing between the genomes it is of considerable interest that the total numbers of SAGs, as well as the representation of different types of SAG families, differs between species. The most pathogenic parasites have relatively high numbers of SAGs and the most immunogenic parasites have relatively low numbers. This may indicate key roles for parasite surface antigens in the outcome of infection, including the degree of damage caused to the gut and the susceptibility of recovered chickens to later parasite challenge.
The data generated from genome sequencing will be invaluable for pursing new cost-effective drugs and vaccines, and also to support longevity of any new products. European and North American poultry production currently leads the global demand for anti-coccidial products, but the biggest future expansion of the poultry industry is set to occur in Africa and Asia, thus new priorities are emerging. Shifts from traditional to intensive poultry production using indigenous and hybrid birds are increasingly common in these regions (Figure 3), although the inherent resistance or susceptibility of these breeds to eimerian infection is unknown. Genome sequences from Eimeria field isolates collected from India and Nigeria will soon be available and comparisons with the reference genome sequences we have generated provides a foundation on which modern molecular biological tools can be used to gain insights into genome evolution, naturally occurring genetic diversity and host/pathogen interactions.
Comments