
Introduction
In 2016, the World Health Organization estimated there were 216 million cases of malaria worldwide. Among them, malaria caused death in approximately 445,000. The emergence of resistance to current antimalarial combination medicines (artemisinin-based combinations or ACT medicines) signifies the importance of identifying novel, anti-malarial drug combinations that can bypass current resistance mechanisms.
Due to the large number of combinations possible, using a computational rationale for optimal selection of compound combinations can save cost and effort by providing a way to prioritize subsets of possible compound combinations. Over the last few years, there have been many efforts to predict the drug responses, with the bulk of such studies focused on oncology. However, there is a pressing need for new drug combinations in other diseases as well (such as malaria) where resistance to current treatment regimens is increasing.
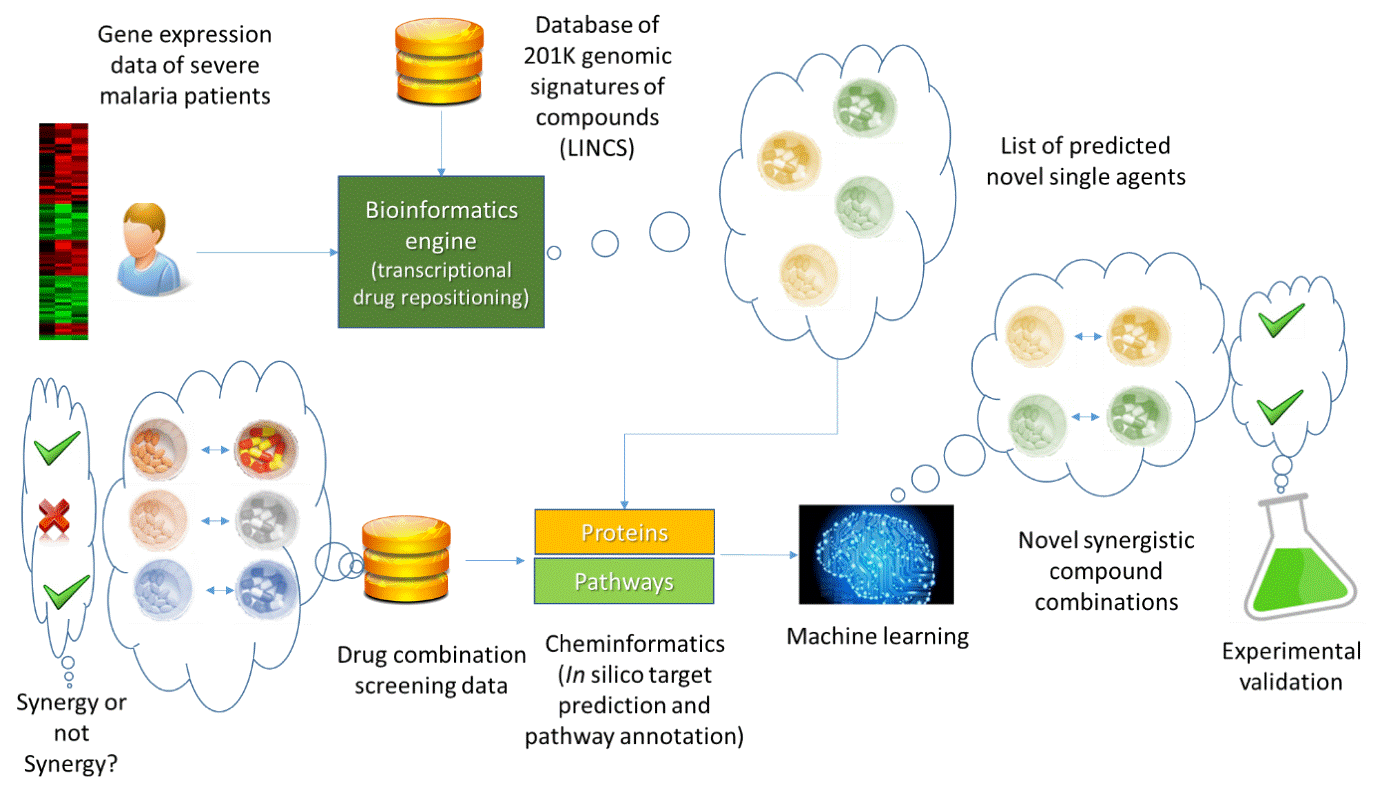
To address this need, scientists from the National Center for Advancing Translational Sciences (NCATS) and the University of Cambridge have developed a computational approach to predicting which small molecule combinations will exhibit synergistic behavior in multiple malarial parasite lines. Synergy occurs when the combined effect of two compounds is superior than the expected result of a single agent (though an additive property). The computational approach that was developed for this study involved bioinformatics, cheminformatics and machine learning techniques, and is depicted in Figure 1:
Methods
The bioinformatics aspect of this study used gene expression data from the blood samples of malaria patients’ with severe cases and compared them to the same patients when they had significantly recovered from the disease. This comparison allowed us to identify key genes that were being differentially expressed in patients who responded to the disease in a curative way. In the next round, a bioinformatics core engine was used that utilized transcriptional drug repositioning for identifying drugs that could stimulate those genes to boost the immunity of patients with malaria.
Through this mechanism, the bioinformatics approach led us to list the single agents that could potentially treat the disease by targeting host genes. However, this was only part of the puzzle as the ultimate goal was identifying which combinations of these single agents could have synergistic effect.
Recently, anti-malarial combination screening data has been generated and is publicly accessible. The wealth of this data enables screening compound combinations in a more informed manner. It also significantly reduces the cost of discovering novel synergistic combinations as only compound pairs which have higher chances of being synergistic are screened.
We utilized the compound screening data of combinations of current and investigational drugs generated at NCATS, and developed a machine learning approach to predict novel synergistic compound pairs. The predictions were then experimentally tested at NIH/NCATS. Notably, this part of the process involves several layers of in-silico target prediction (cheminformatics) and pathway annotations to be able to translate the effect of compounds in a series of numbers to the machine learning engine.
Results
The performance of the algorithm to determine mild to strong synergies was impressive, with 83% of the compound pairs that were predicted to be synergistic were experimentally validated. The model was able to detect 65% of the synergistic pairs that were found experimentally. Of course, it is harder to identify highly synergistic pairs due to the fewer examples of such cases in the NCATS dataset. The percentage of experimentally validated predictions and the detection rate of experimentally valid synergies were 49% and 76% for moderate-to-strong, and 12% and 67% for strong synergies, respectively.
One of the single agents identified from the boinformatics part of the approach was Apicidin. This compound is a fungal metabolite as well as a histone deacetylase inhibitor and at the dose which is safe for humans (500nM) can kill on average 97% of the parasites. Apicidin has not yet been approved for malaria treatment, and requires further clinical studies to pass safety and efficacy tests in patients. Our approach was also successful in identifying novel, strongly synergistic pairs of compounds such as Tacrolimus-Hydroxyzine and Raloxifene-Thioridazine. These compound pairs exhibited significant synergy across three P. falciparum strains (3D7, DD2 and HB3) included in the study.
Conclusion
This work highlights the ability of bioinformatics, cheminformatics and machine learning methods to identify synergistic drug combinations for malaria, using a combination of genomic and pharmacological data as inputs. The performance of the resulting model is suitable for the prioritization of potential compound combinations in an informed manner. Notably, the computational approach is conceptually also applicable to other infectious diseases. One potential future study for this algorithm can be its application in the field of precision medicine and finding synergistic anti-malarial pairs based on the genomics data analysis of patients that have developed resistance to current therapies.
This work was a successful example of how computational approaches are transforming drug discovery by accelerating the process and ultimately reducing the cost of drug development. These fields require further attention and an increase in collaboration between computational scientists and biologists to make the best use of computational power and bridge the gap between fields. Multidisciplinary studies like ours contribute towards making life-saving discoveries, and should be a common practice in the future.
Comments