
The complexity of cancer has famously eluded conquering by modern medicine. Every tumor has many aberrations that drive its growth. As a result, treatments that target single vulnerabilities are typically of short-lived efficacy. After being diagnosed with advanced stage ovarian cancer in 2013, I wagered that what was needed was an algorithm capable of digesting and analyzing the complexity to provide a detailed view into the multitude of factors at work in a given tumor.
To pursue this goal, I began a collaboration with Greg Ver Steeg, who specializes in analyzing big data, to bring state-of-the-art machine learning to bear on the recently released large-scale data from the Cancer Genome Atlas (TCGA). TCGA contains publicly available comprehensive maps of the key genomic changes in 33 types of cancer.
Finding patterns
Greg previously developed a machine learning method called Correlation Explanation (CorEx) which we applied to TCGA tumor data. CorEx uses information-theoretic principles to find hidden factors that ‘explain’ relationships in the data. In our case, these factors account for dependencies (correlations) among tumor genes.
During our initial discussions, it was clear that CorEx showed promise, but required refinement to squeeze as much information as possible out of noisy gene expression data. Subsequent innovations in this context allow CorEx to learn patterns efficiently from relatively small numbers of patient genomic profiles.
Greg and I used the improved CorEx to find patterns in RNA-seq gene expression data for 420 ovarian tumors. CorEx was able to find an extraordinary amount of structure in the data, much of which was associated with known cellular functions and pathways. We identified genes whose expression seems to be linked in ovarian cancer and with all these expression dependencies mapped out, we could begin to ask how they fit into a larger framework for understanding of tumor biology and treatment.
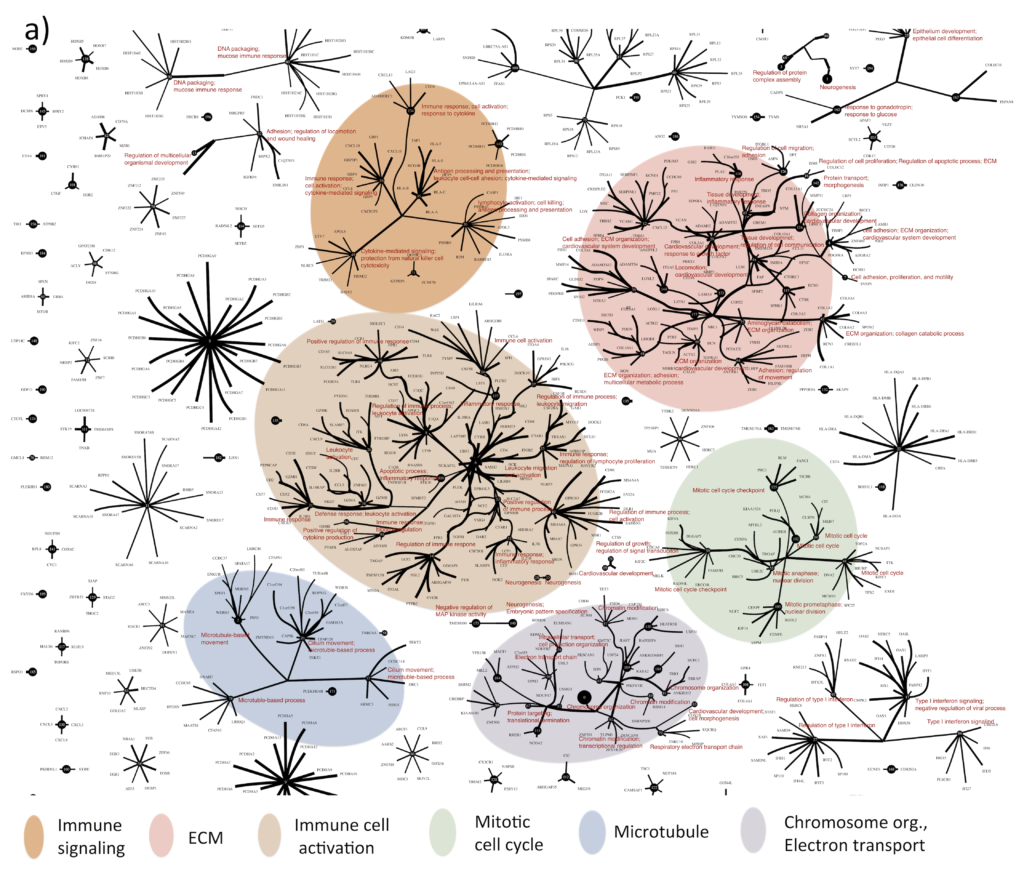
Towards targeted treatment
One of the questions we asked was how to combine treatments in order to extend patient survival. We were able to show that combinations of the CorEx factors (i.e. patterns of gene expression that CorEx identified) were significantly associated with survival among the TCGA patients. This suggests a method for selecting combination therapies based on these patterns for future clinical trials.
We also asked whether any factors associate with long term survival (a particular concern for me!). One specific factor stood out as a candidate. It contained several proteins regulating stemness properties – such as the ability of cells to self-renew and differentiate into different cell types – that are implicated in aggressive metastatic disease and chemoresistance.
Our analysis shows that tumors containing many cells with stemlike gene expression correlate with poor long-term patient survival. CorEx is especially good at detecting weak correlations in large sets of variables, and this is likely why it was able to detect this particular pattern for the first time in ovarian cancer expression data.
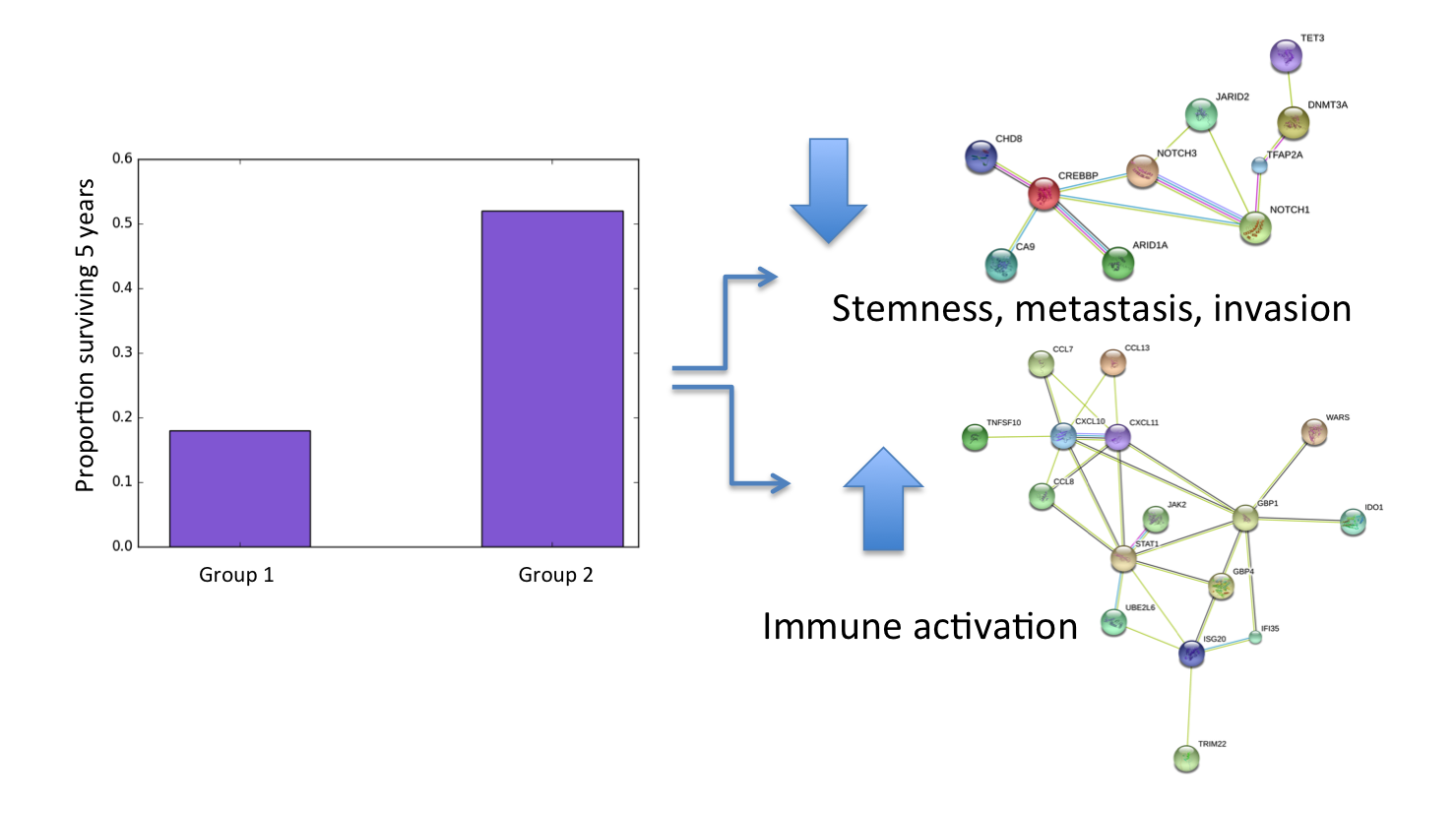
While these are interesting general highlights, the real significance of our findings lies in CoreEx’s capacity to identify multiple target networks in an individual tumor. When I recurred less than a year after completing my initial chemotherapy, I was able to compare my tumor’s gene expression to patterns of gene expression CorEx had identified in the TCGA data. I noticed that the expression factors present in my tumor suggested a very favorable immune activation. Generally, this would predict a favorable response to chemotherapy, but unfortunately that had not been the initial outcome.
I used this information from CorEx to select an unconventional second chemotherapy regimen including an immune checkpoint inhibitor, which allows immune cells to recognize and attack cancer cells.
Greg and my efforts have borne fruit: I am still alive and off therapy for 18+ months – a second remission that is at least twice as long as the first one, which is rare in recurrent ovarian cancer.
While we cannot be sure that the extended remission was due to my algorithm-driven treatment choices, what is certain is that some patients benefit greatly from personalized treatment options and there is a great need to expand this benefit to more patients.
Our analysis of the TCGA ovarian cancer RNA-seq presents a rich tableau of tumor biology, of which we have only scratched the surface. We expect deeper exploration of the various factors that drive individual tumor growth, spread and survival will yield further insights to inform more rational, targeted treatment selection. Although much further research is needed before this method can be widely applied, with an algorithm like CorEx that is capable of sorting out the daunting complexity of tumor cell biology, we are hopeful the tide will soon turn in more cancer patients’ favor.
Comments