
Alzheimer’s disease (AD) is a highly heterogeneous disease, caused by multiple known and unknown factors, it can therefore be extremely difficult to develop treatments by targeting specific causes for individual cases. In the majority of AD cases memory deficits, in particular those associated with consolidating short term memory, occur in different forms at different stages of the disease. A study published in Molecular Brain reports on a potential therapeutic tool to correct synaptic signaling and restore memory in Alzheimer’s disease mice models through PKR inhibition.
But how does memory consolidation work?
Neural plasticity is an umbrella term used to describe a permanent change to the brain. These occur throughout people’s lives based on the persistent use, or lack of use as the case may be, of certain neuron pathways. Neural plasticity at the synapse level can take the form of long term potentiation (LTP) or long term depression.
Strengthening synapses
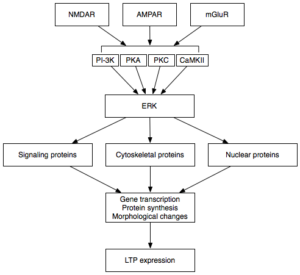
LTP is the form of plasticity responsible for the conversion of short term memories to long term memories, known as memory consolidation, through the strengthening of specific synapses used. Upon chemical activation of the post-synaptic neuron a molecular cascade is triggered:
- Protein kinases assist in the autophosphorylation of RNA activated protein kinases (PKRs).
- Once phosphorylated these activated PKRs can move into the nucleus and activate transcription factors within the nucleus.
- Transcription factors produce molecules which can either aid or suppress LTP.
In AD, eIF2α is of particular importance. This transcription factor, found in the position of ERK in the flow diagram, is responsible for producing molecules whose role is the suppression of memory formation. These inhibit transcription of molecules essential for LTP, such as those involved in reception production and synapse strengthening.
In the post-mortem brains of AD patients and AD mouse models, PKRs and eIF2α are found to be hyperphosphorylated, meaning these molecules show higher phosphorylation levels than normal. This excessive phosphorylation will result in elevated activation and overproduction of LTP suppression molecules resulting in synaptic dysfunction. Targeting excess PKR activation may provide a potential therapeutic tool in restoring memory deficits in AD.
How can we restore these LTP deficits?
Model Selection
Researchers selected two mouse models to use.
The 5xFAD transgenic mouse line provided a model which recapitulates rapidly developing severe amyloid pathology. This is accompanied by synapse degeneration, reduced neuronal firing, neuronal loss and spatial learning deficits with LTP impairment at around 6 months of age.
The other was generated through the injection of Aβ1-42 oligomers in ICR mice allowing investigations as to whether PKR inhibition works in multiple AD mouse models.
Researchers used the method of inhibiting the activation of PKRs in order to restore LTP as it prevents the production of inhibitory molecules.
The outcomes
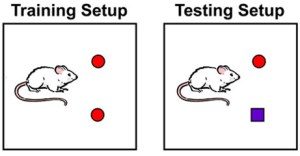
Behavioural tests, namely a novel object recognition task, were carried out on the Aβ1-42 ICR model. 2 days after their Aβ1-42 oligomer infusion the mice were trained on the novel object recognition task which was repeats 24 hours later to test their long term memory. The mice displayed defective memory consolidation in this case.
Application of the PKR inhibition pre training significantly improved the long term memory deficits seen previously increasing the mice’s preference index for the novel object. This suggests a positive effect on the mechanisms of LTP even in mice that were a year old with advanced disease.
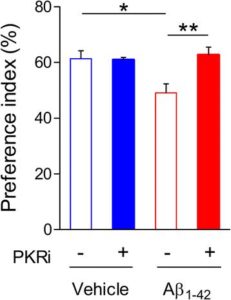
This LTP recovery was also seen in tests on hippocampal slices from both models when they received treatment 30 minutes prior to and 30 minutes after LTP induction.
But what implications could these findings have on future AD research?
It still remains slightly unclear as to the molecular mechanisms behind this treatment. Western blot analysis carried out showed a reduction in the phosphorylation levels of both PKR and eIF2α suggesting that inhibition treatment restores the phosphorylation levels to that seen in the normal human brain. However these reductions weren’t statistically significant.
Recently there has been numerous reports on failures of clinical targets targeting the cause of the disease β-amyloid. Therefore pharmacological interventions targeting neural plasticity may provide a promising alternative strategy for the development of AD treatments. These interventions would disregard the individual etiology of a patient’s disease and target a common symptom. The success of PKRi on both old and young mice, and its lack of effects on Aβ seen in 5xFAD mouse models, suggests that this treatment may provide a new target suitable for both early and late stage AD to alleviate memory loss symptoms.
- The new protein on the learning and memory scene: Sirtuin 6 - 31st October 2018
- Autism Awareness Week Quiz 2018 - 26th March 2018
- Ghrelin: a new therapeutic target for Parkinson’s? - 20th February 2018
Comments