It is ten years since Dr Shinya Yamanaka reported the Nobel Prize winning technology of induced Pluripotent Stem Cells (iPSCs). In addition to its significance in reprogramming of somatic cells to become pluripotent, there is still an increasing interest in iPSC-technologies, including the application to medical science and the development of new therapeutic interventions, using cell based regenerative medicine and disease modeling.
My colleagues and I have been heavily involved in disease modeling of neurological and psychiatric disorders using iPSC-technologies. We have established iPSCs from more than 30 neurological and psychiatric disorders. These can be classified into pediatric neurological diseases, hereditary diseases affecting sensory organs, mental disorders and late-onset neurodegenerative disorders. In our article in Molecular Brain, we reported on the characterization of lissencephaly, a severe pediatric neurological disease.
What is lissencephaly?
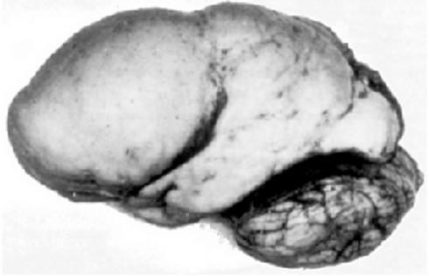
Lissencephaly is one of the most severe congenital brain malformations, that shows disturbed cerebral cortical lamination. The name ‘lissencephaly’ means ‘smooth brain’, since the surface of the brain appears smooth owing to insufficient gyration.
It has been postulated that neuronal migration in the developing cerebral cortex is impaired in lissencephaly, based on the mouse models and human genetics. Several genes involved in microtubule regulation, such as LIS1, DCX, ARX and TUBA1A, are known to be mutated in human lissencephaly.
The molecular and cellular etiology of human lissencephaly has not been experimentally investigated due to its rareness and early postnatal fatality
Thus, it is likely that dys-regulation of microtubule dynamics lead to the impaired neuronal migration in this disease. However, the molecular and cellular etiology of human lissencephaly has not been experimentally investigated due to its rareness and early postnatal fatality.
What did we do?
To overcome this difficulty, we have established the iPSCs from lissencephaly patients with a missense mutation in TUBA1A, encoding a subtype of alpha tubulin, which is highly expressed in neural progenitor cells and young neurons.
We characterized iPSCs from two patients of TUBA1A gene mutations, i.e., R264C and N329S mutations, which are likely to affect alpha tubulin protein’s interaction with Dcx protein and reduce the number of hydrogen bonds between alpha- and beta-tubulin, respectively.
To characterize the possible phenotypes that are relevant to disturbed cerebral cortical lamination in vitro, we used our previously developed method, the glia-supported neurite extension method. Translocation-like movements of neural progenitor cells (or young neurons) can be observed in this method; thus, we expect that our co-culture system might be useful in the neuronal migration of iPSC-derived telencephalic neurons.
What did we find?
We found that neurite extension was severely disturbed in the neural progenitor cells differentiated from a severe lissencephaly patient-derived iPSCs in this glia-supported neurite extension assay.
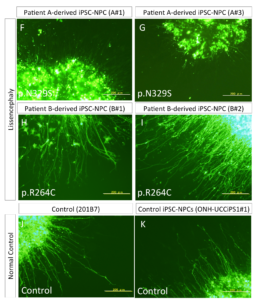
The measurement of neurite extension after 5 days showed that neurite extension in iPSC-NPCs derived from the patient with the p.N329S TUBA1A mutation was severely inhibited (Figure 4 F–G and L), This neural extension inhibition was observed less in patient B (Figure 4 H-K) which was consistent with the gradient in the pathology along the cephalic-caudal axis, observed in the patients.
These data suggest neuronal cells that differentiated from the iPSC-NPCs with the p.N329S TUBA1A mutation had an immature morphology compared with those derived from the control iPSC-NPCs. We believe that this phenotype corresponded to the pathology of the developing brain because neuronal cells initially polarize from NPCs and migrate toward the cortical plate using various types of neurites, including the leading process and basal process.
This is the first report of the characterization of neurites extension in human lissencephaly and it could be considered that lissencephaly-iPSCs would provide the clue to understand the role of microtubule in the pathogenesis of lissencephaly and the mechanism of human cortical development.
Modeling human disease like lissencephaly is becoming feasible using a special culture method that mimics a part of organogenesis in combination with patients-derived iPSCs
Collectively, modeling human disease with a defective organ development like lissencephaly is becoming feasible using a special culture method that mimics a part of organogenesis in combination with patients-derived iPSCs.
In other words, further development of human iPSCs-derived 3D organoid culture technology would enormously contribute to understanding of human development and diseases.
Comments