The last decade has seen massive improvements in genomic sequencing and assembly and by now researchers are able to obtain nearly perfect genome sequences for bacteria and small eukaryotes in short time and at relatively low cost. Making good assemblies on the cheap possible even for small research labs enabled a democratization of sequencing and resulted in many new draft genomes, including various new parasite genomes. This trend is reflected in an increasing number of available tools for sequence assembly (>60 by 2016).
However, the problem of generating high quality standardized annotations for these organisms, i.e. locations and functions of genes and other relevant features, remains. The availability of detailed and complete annotations is key to enable subsequent comparative cross-species analyses to identify differences between individual species or strains.
Examples for such differences could be loss or gain of common and/or species-specific genes and functions. In the bacterial world, software tools to quickly annotate genomes exist, but up to now an equivalent for parasites was missing.
But even if a draft assembly is available, without annotation it remains an incomprehensible string of data without scientific meaning.
Introducing a new software tool
As a response to this need we developed Companion, a new software tool and web server to generate comprehensive annotations of parasite genomes in very short time, making use of information we already know about related species.
Companion’s unique features include visualization of assembly quality, comparison of gene content to the reference genome as well as delivering files that can be easily submitted to public databases like the European Nucleotide Archive (ENA).
Giving data a scientific meaning
But even if a draft assembly is available, without annotation it remains an incomprehensible string of data without scientific meaning. To make any use of it, one needs to know the locations of protein-coding and non-coding genes, and what their functions are.
Although finding these is an old challenge for which specific tools are available, the task of gene finding is still an open problem. Less than perfect annotations, i.e. missing, wrongly or only partially described gene models as well as erroneous function associations, might severely impact any kind of downstream analysis.
However, reality has shown that best results are only obtained by the use of multiple tools in parallel, followed by manual curation.
Finally, while a requirement for publication, the submission of annotation files to databases like the ENA is usually a challenge, as specific nomenclature must be followed.
How can Companion be used?
To help the parasite community to overcome those problems we have developed the Companion (COMprehensive Parasite ANnotatION) software as a free resource for public use. Though primarily available as a web server, it can also be installed locally to annotate genomes that could not be run online.
For the main target audience of parasitologists, we provide previously unmatched simplicity of annotation: just upload the assembly, select a related reference species from our set of 62 parasite genomes, and press a button.
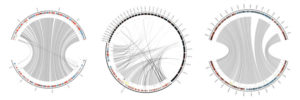
After 4-6 hours (depending on assembly quality and reference size), an email is sent directing the user to their annotated genome. Companion provides basic statistics such as, number of genes, gene density, proportion of each T, C, G and A base in the DNA etc., but also – more interestingly – first comparative results such as a phylogenetic tree that illustrates the newly annotated species’ relation to other species, or the gene content relative to the reference. Also the quality of the assembly, as well as large-scale rearrangements, is easily observed in the automatically generated circular plots. If the user is happy with the result, it can now easily be uploaded to the ENA, a process that used to be another big effort in the past. Of course the annotation generated by Companion can also serve as a good starting point for subsequent manual curation.
Putting Companion to use
The first major use case was using Companion to annotate various new kinetoplastid genomes, including 12 Leishmania as well as Trypanosoma, along with Crithidia and Endotrypanum genomes, most of which are available from TriTrypDB.
Companion is generating a high quality draft annotation that can easily be submitted to the databases to enable the community to learn from those sequenced parasites.
Since Companion’s public launch in early 2016 we have counted over 120 annotation runs from all over the world, with increasing popularity. At this point we would like to thank the Wellcome Trust Sanger Institute infrastructure systems team for maintaining the server. Companion has proven to be versatile: even though the main purpose of Companion is to annotate whole genomes, users have reported that they sometimes only use it for the pseudochromosome contiguation component, a functionality that is also scarce as a web application.
Companion is implemented using state-of-the-art technology: the Nextflow workflow management system to orchestrate the pipeline, the GenomeTools genome analysis toolkit for low-level scripting, and the Rails development ecosystem for the web server. All code is available under a free open source license.
In conclusion, companion is generating a high quality draft annotation that can easily be submitted to the databases to enable the community to learn from those sequenced parasites. It also provides various outputs that allow the user to finally compare the newly annotated genome to the reference, possibly leading to first directions for further research.
More information can be found on GitHub. The related paper was recently accepted in Nucleic Acid Research’s web server issue (PMID: 27105845). Current improvement of the software will focus on extending its use to fungal genomes.
Further links:
• Companion application: https://companion.sanger.ac.uk
• Companion software: https://github.com/sanger-pathogens/companion
• Mathew Berriman’s Parasite Genomics group at the Sanger Institute https://www.sanger.ac.uk/science/groups/berriman-group
• Mathew Berriman’s website at the Sanger Institute https://www.sanger.ac.uk/people/directory/berriman-matt
• Thomas D. Otto’s website at the Sanger Institute https://www.sanger.ac.uk/people/directory/otto-thomas-d
• Sascha Steinbiss’s website at the Sanger Institute https://www.sanger.ac.uk/people/directory/steinbiss-sascha
Comments