Imagine that you are trying to maintain a bridge made from two types of timber which must be procured from two different timber mills, and painted in a custom color that was mixed at a paint store.
You can either visit each timber mill and the paint store to obtain the material you need each time your bridge needs fixing, or you can stock pile the timber and paint until it is needed. If you visit the stores every time you need new supplies, you don’t have to waste space on timber and paint.
However, it’s a hassle to always have to visit each of these sources. If you stockpile the timber and paint, you won’t have to drive to each source every time, but your paint and timber can be unusable over time due to the paint drying or the timber warping.
You struggle with these two issues to maintain the bridge until a new hardware superstore opens up. The superstore carries both types of timber, can mix the correct paint color for you–and they have free shipping. Suddenly, maintaining the bridge becomes easier so you can spend your time doing something else.
Maintaining a pipeline of information
Now imagine that instead of three physical components from three different locations, you have hundreds or thousands of virtual components from seven to fifteen data sources—that each of these data sources provide the components in a slightly different fashion, and that instead of a physical bridge, you are maintaining a pipeline of information.
Vast swaths of gene and variant annotation information are spread across many different resources, making it challenging for researchers
Welcome to the big data landscape of gene and variant information! Vast swaths of gene and variant annotation information are spread across many different resources, making it challenging for researchers to integrate up-to-date information into their bioinformatics pipelines.
Researchers typically address this challenge with data warehousing or data federation. By downloading and storing data from different resources (data warehousing), researchers ensure fast access to data of interest to them; however, effort must be spent on writing papers and keeping the data up-to-date.
In contrast, by accessing the data directly from the resource when it is needed (data federation), researchers ensure that they obtain the most up-to-date information available from these resources, but may find their queries to be time consuming due to server and network limitations.
An alternative solution
In our recently published paper in Genome Biology Jiwen Xin, et al. describe an alternative solution for obtaining up-to-date gene and variant annotation data from multiple resources: the annotation as a service.
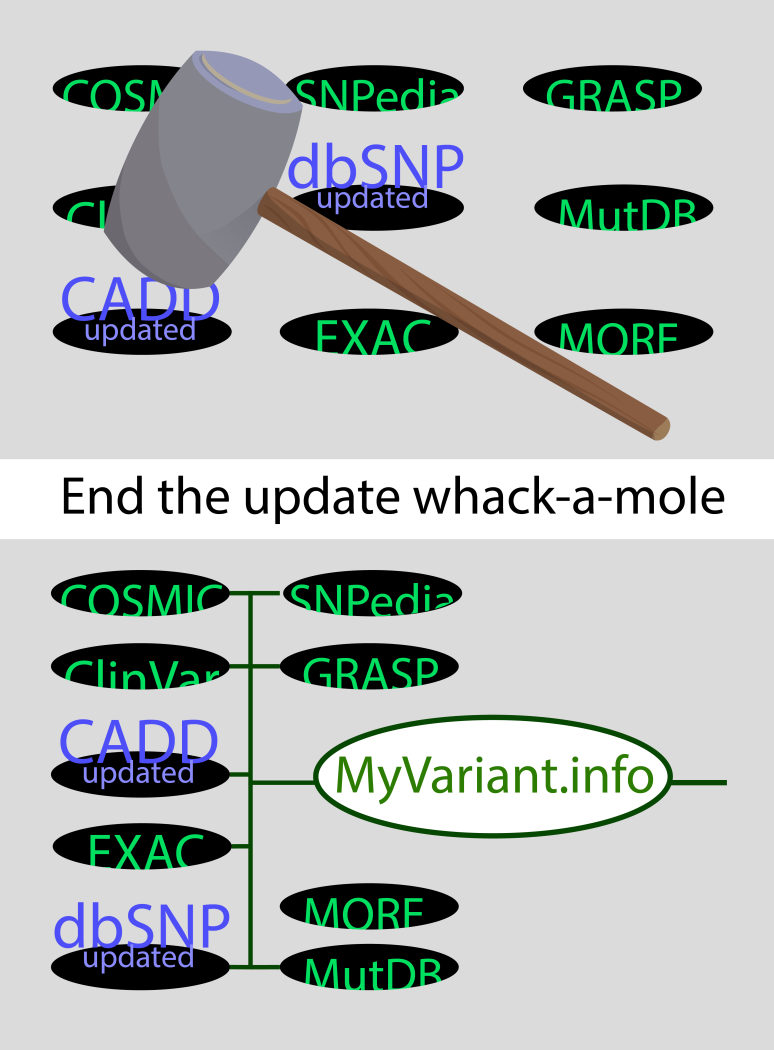
Like the hardware superstore in bridge example, MyGene.info and MyVariant.info are one-stop shops (i.e. centralized repositories) that serve up-to-date annotation data from key resources via cloud-based web-service endpoints.
MyGene.info stores up-to-date data from NCBI Entrez, Ensembl, Uniprot, NetAffy, PharmGKB, UCSC, and CPDB.
Instead of dealing with data warehousing or data federation issues in addition to data format conversion from multiple data sources, researchers or bioinformaticians can utilize any of MyGene.info’s clients (Python, R) or browser-based API to access up-to-date gene annotation data in a single machine-readable format (json).
For example, MyGene.info can easily be used to batch convert gene IDs or obtain pertinent gene ontology info—two tasks for which researchers commonly use and cite DAVID. Providing easy access to gene annotation information like gene IDs and gene ontology is so valuable that researchers continue to use DAVID for this purpose even though DAVID has not been updated for a long time!
A valuable resource and solution
After confirming that researchers would find this resource valuable and seeing the volume of requests we get monthly, we wanted to find a similar solution for gene variant annotation data. That was the idea behind MyVariant.info
“With over 50 different annotations types covering over 13 million genes for 15,000 species, MyGene.info has already accumulated over 160 million requests, and serves an average of 3.5 million requests per month!” Dr. Chunlei Wu revealed, the Associate Professor at the Scripps Research Institute in charge of developing these services.
Elaborating on the development of MyVariant.info, he added, “After confirming that researchers would find this resource valuable and seeing the volume of requests we get monthly, we wanted to find a similar solution for gene variant annotation data. That was the idea behind MyVariant.info.”
MyVariant.info currently incorporates up-to-date variant annotation data from fourteen valuable resources including: dbNSFP, dbSNP, ClinVar, EVS, CADD, MutDB, GWAS Catalog, COSMIC, DOCM, SNPedia, EMVClass, Scripps Wellderly, EXAC, and GRASP.
As a service aimed primarily at bioinformaticians and developers of bioinformatics tools or resources, MyGene.info has already been incorporated into public resources such as BioGPS, the Monarch Initiative, and CIViC. BioGPS provides about 40% of the traffic to MyGene.info, while 60% of the traffic comes from over 5000 unique IP addresses.
Both MyGene.info and MyVariant.info are open source projects. Links to the source code, use case examples, and the clients can be found in the original paper in Genome Biology, here.
- Tools to annotate genes and genetic variants - 25th May 2016
Comments