
Read mapping is the process of aligning short sequences (also known as ‘reads’) to a reference genome or a de novo assembly. It is a major analysis step in nearly every Next Generation Sequencing (NGS) related experiment (single nucleotide polymorphism (SNP) calling, RNA-seq, ChIP-seq, and so on), and is performed daily by several thousands of researchers all over the world.
Identifying the right region
The main problem of mapping is to identify the region on the genome where a given read most likely originated. At the beginning of my PhD, I thought that this is a seemingly straightforward problem. However, considering all the possible genomic variations, as well as PCR or sequencing artefacts, and solving issues like repeating regions is challenging.
Current research in this area has mainly focused on speeding up or reducing the memory requirements, and there are around 100 mappers (programs to align reads) that have been published.
To choose a mapper for a given study from this long list, others and I often rely on word-of-mouth recommendation, on the number of citations, or on a few benchmark papers. However, this is hardly a rigorous process, and a mapper used in one study might not be the optimal choice, or even a good choice, for another.
Every read is important and not mapping even 0.1% of your reads might lead to inaccurate results.
Furthermore, choosing the optimal parameter settings is advantageous over using the default settings that are most often optimized for speed. Without this optimization, important SNPs might be missed, gene expression could be misevaluated, or any number of other possible artefacts might be introduced.
Every read is important and not mapping even 0.1% of your reads might lead to inaccurate results. However, often not even the developer of the mapper can predict the best parameter set for a given data set.
Introducing Teaser
In our recent article in Genome Biology, I introduced a new website and command line program Teaser that addresses these problems by automatically benchmarking mappers and their parameter settings.
My main motivation was to make it fast and simple to evaluate how different mappers perform with your datasets. For example, benchmarking six different mappers on a human genome can be done over a coffee break (~15 minutes).
On your return, you will be provided with interactive plots that visualize how well each mapper performed based on the percentage of reads correctly mapped and its throughput (reads per second).
In addition, Teaser should be easy to run, but still tuneable and customizable for everybody. This includes the biological conditions of the experiment such as the particular genome sequence and rate of heterozygosity as well as experimental conditions like the read lengths or error rates.
Once these conditions are listed, Teaser can automatically run any number of candidate mappers and parameter settings of interest. This includes trying an ensemble of parameters, such as trying different seed length while varying two additional parameters to increase the attempts to align reads.
Now it is possible that 34 tests that are run automatically within 20 minutes (a coffee plus a cookie) on a Drosophila melanogaster data set. Doing this manually will occupy a student for almost one month.
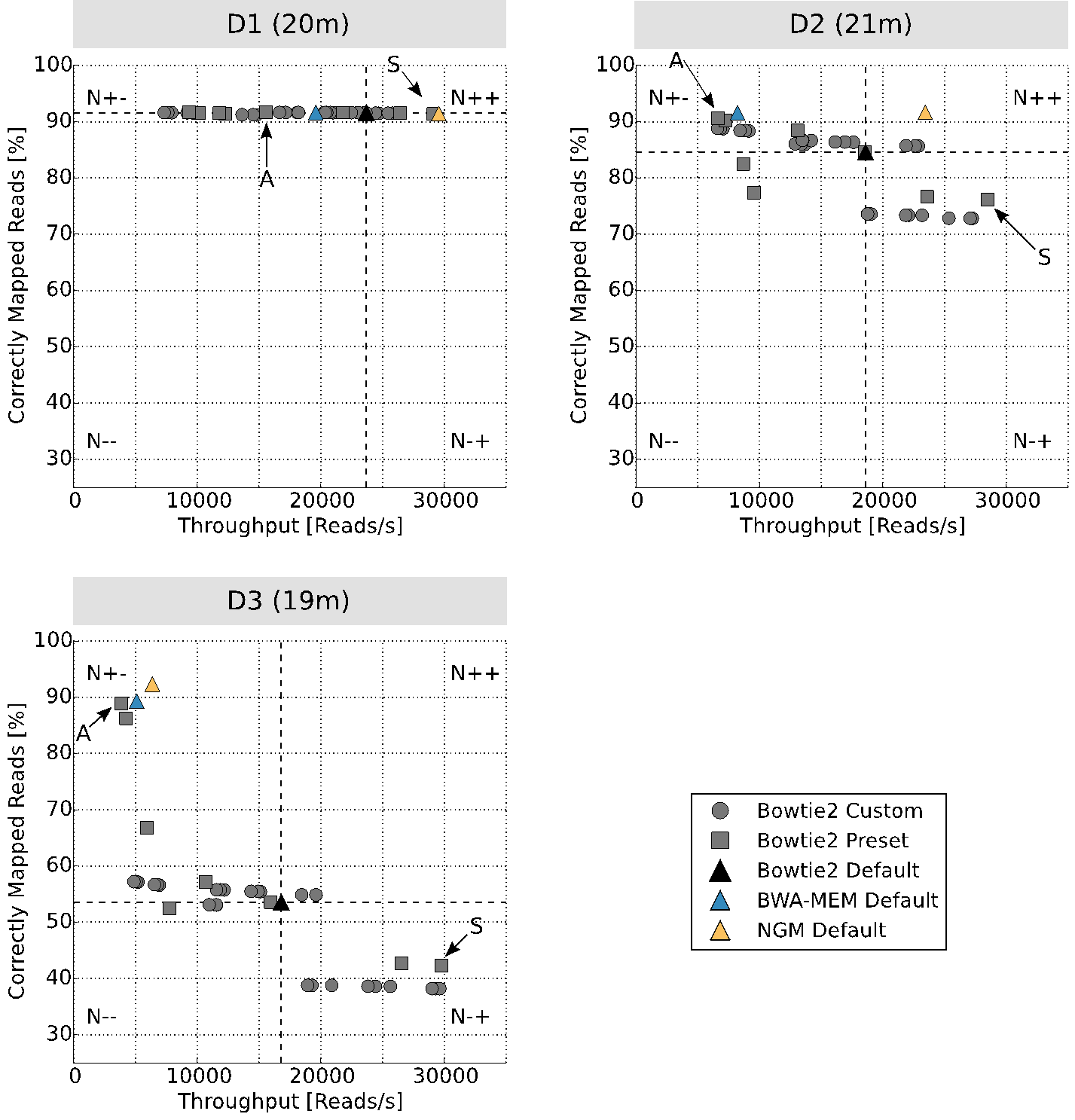
The image above shows the result. You can choose your mapper/parameter setting from four regions with respect to running the default parameters of, for example, Bowtie2: lower left (N–) are the result that are worse than the default, upper left (N+-) are the results with higher mapping rate but longer runtime, lower right (N-+) are the result with lower mapping rate and lower runtime and (N++) are the results with higher mapping rate and lower runtime. I personally always choose settings from N+- or N++, depending on the goal of the study.
To summarize, Teaser can assist in choosing the optimal mapper and parameter settings for your data. This is important for every biologist that has invested thousands of dollars to get his or her sequencing library and now wants to utilize the reads as much as possible.
Furthermore, Teaser can justify the usage of a mapper and parameter setting in a study, especially if you are analysing a new genome or trying a new read type. In addition, Teaser is also helpful for developers like me as it eases the benchmarking over novel mappers.
Teaser is available as a stand-alone version as well as a web application.
- Introducing Teaser: Optimization of read mapping results over a coffee break - 28th October 2015
Comments