Genomic surveillance is commonly used to track viral and bacterial pathogens, such as identifying new variants of SARS-CoV-2 and linking outbreaks of food-borne disease back to particular products and geographic areas. Similar tools can accelerate progress towards the elimination of endemic neglected tropical diseases like schistosomiasis but have historically been harder to develop and use for such multicellular parasites. Parasites have larger genomes and more complex life cycles than viruses and bacteria, making it more technically challenging and expensive to sequence and analyze their DNA at scale. But recent advances in sequencing technologies have made whole genome sequencing increasingly affordable for population-based studies of schistosomes and other complex parasites. These advances create new opportunities to address key threats to schistosomiasis control and tailor interventions to local contexts.
Threats to schistosomiasis control
Over the past two decades, schistosomiasis control efforts have focused on expanding treatment coverage through mass drug administration (MDA). These programs have reduced morbidity and infection intensity but have not achieved the World Health Organization’s (WHO) control and elimination targets. In fact, schistosomiasis still affects 200 million people globally, causing disease and disability that disrupts physical growth, cognitive development, and labor productivity. Its transmission between definitive vertebrate hosts and intermediate snail hosts persists in some areas despite treatment programs and has reemerged in other areas where it was previously controlled. We do not yet fully understand why this is. It could be that animals play an underappreciated role in the transmission cycle, that the movement of people re-introduces the parasite into places where control efforts are ongoing or that MDA campaigns are not reaching enough people to interrupt transmission. We need new tools to fill these gaps in our understanding and address how different social and environmental processes can undermine MDA programs and threaten their success. A recent review paper outlines how whole genome approaches can reveal mechanisms behind persistence and reemergence of schistosomiasis. In it, we describe two key threats to schistosomiasis control, what we know about them and how the thoughtful use of genomic data can inform which interventions to implement alongside MDA in different contexts.
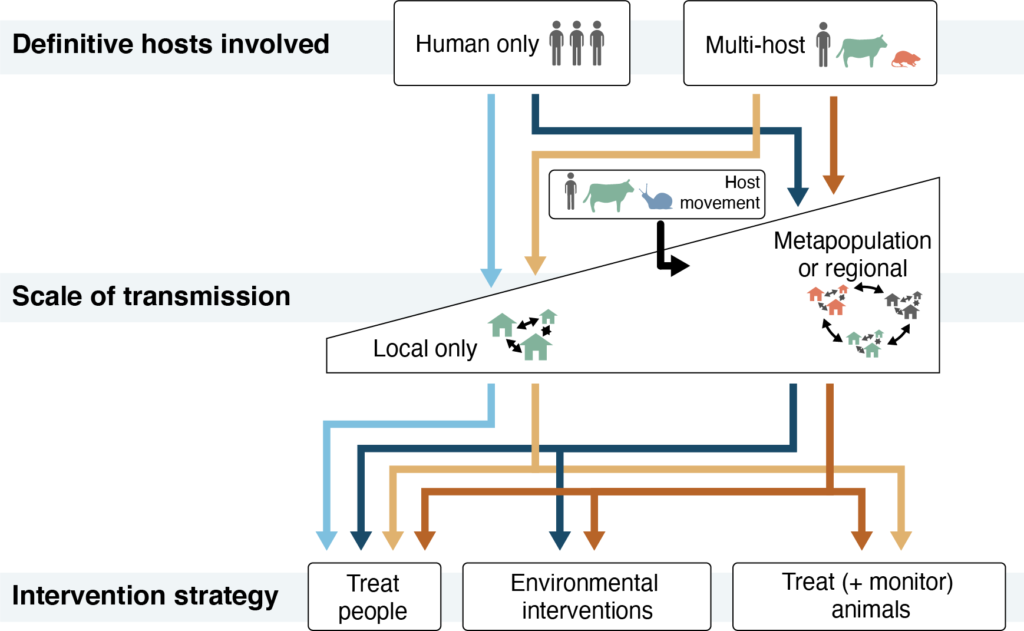
These threats – the contributions of non-human definitive host species to human infection and the importance of locally-acquired versus imported infections – are key to understanding the contexts in which MDA is sufficient and when additional interventions are necessary. We argue that MDA programs are likely to be sufficient only when infections are found primarily in people (with limited involvement of non-human definitive hosts) and are acquired locally (rather than imported from elsewhere). In contrast, frequent involvement of non-human definitive hosts, such as livestock or wildlife, may undermine MDA programs by allowing parasites to persist in species that are not the targets of control programs. This can be addressed by monitoring and treating relevant animal species. Similarly, the movement of infected people, animals, and snail hosts can threaten the success of MDA by introducing parasites from outside a program’s catchment area. This threat can be mitigated by supplementing MDA with environmental interventions (such as snail control and the provision water and sanitation infrastructure) to reduce exposure and contamination and the risk of re-infection after treatment.
Zoonotic transmission of schistosomes
Historically, questions related to the contributions of non-human definitive hosts and parasite importation in the transmission of schistosomiasis have been addressed with a variety of epidemiologic, ecological, and genetic methods. This work has revealed how the role of non-human definitive hosts varies across schistosome species and geographic contexts and how parasite importation can create new foci of transmission as well as sustain existing foci of transmission, even in the presence of control efforts. It has also revealed that closely related schistosome species in the Schistosoma haematobium complex, which infect humans as well as livestock, can mate with each other and form viable hybrids, potentially complicating control efforts. The genetic tools used in these past studies most often used microsatellites (MSATs), short repeated sequences of non-coding DNA that mutate rapidly and the sequencing of genes found in the mitochondria (mtDNA) and ribosomes (rDNA) of eukaryotic cells, which are easy to amplify and sequence because they occur in high numbers.
Despite the widespread adoption of MSATs and mtDNA/rDNA markers in genetic studies of schistosomes, they are limited in the information they can provide. MSAT studies can identify approximate patterns of relatedness but because they often include just a few small sections of non-coding DNA (representing a small portion of the genome), they do not reveal much about how parasites are adapting to their environments. It is also difficult to replicate and compare MSAT analyses across laboratories. Similarly, mtDNA/rDNA markers can be used for “bar-coding” or identifying which schistosome species is present, but they have relatively low levels of variation, which limits their ability to detect relatedness between parasites or how they respond to control efforts or host availability in their environments. Whole genome sequencing generates more information, allows for inference of relatedness at fine scales and does so with workflows that are generally more reproducible.
Leveraging whole genome approach for schistosomiasis control
Whole genome approaches can overcome the limitations of older approaches in addressing both the contributions of non-human definitive hosts and parasite importation to schistosome transmission and risk. They can be used to identify transmission events between species (e.g., non-human to snail to human), and determine whether they occurred in the past, are ongoing or are shifting over time. This may be particularly important in low-transmission contexts, such as China, where control efforts that reduce prevalence in certain target species (e.g., humans) may increase the importance of other, non-target species (e.g., rodents) as definitive hosts. Details about the frequency and timing of hybridization events between schistosomes can also be inferred using whole genome approaches, as can the geographic origins and destinations of parasite introductions driven by the movement of human, animal, and snail hosts.
Integrating whole genome data with epidemiologic and ecological data can have an immediate impact on our scientific understanding of key threats to schistosomiasis control. The knowledge generated by these advances can support decisions about which interventions to implement alongside MDA in different contexts. But substantial challenges remain to integrating genomic workflows into surveillance systems. Using genomic data in surveillance systems on time scales relevant for operational decision-making will require investment in resources and local infrastructure in endemic regions. The good news is that costs are declining for whole genome approaches and their replicability across laboratories creates opportunities for standardization and curation of publicly available resources. It is our hope that epidemiologists and geneticists will expand collaborations to harness these increasingly available technologies to tackle the issues of schistosomiasis persistence and reemergence and accelerate progress towards elimination.

Comments