We take a look back at recent developments in the fast-paced field of metagenomics – and look forward to what the future has in store
The term metagenomics first appeared in a publication in 1998 and according to Wikipedia ‘…is the study of metagenomes, genetic material recovered directly from environmental samples‘, and involves the sequencing of many individual microorganism genomes from those samples. This is in contrast to clonal (single, pure) microbial cultures which are commonly used for sequencing in microbial genomics and microbiology.
Metagenomics is an increasingly important area of research in genomics and other related fields. Diverse applications and new software developments continue to be made to improve the identification of mixed cultures of micro-organisms in both unusual and common environments. Much of the sequence data has been made publicly available.
Published data sets include those hosted at EBI; which has a ‘Long insert human faecal metagenomic library’ and a ‘Metagenomic analysis of Ruminal Microbes’ in its catalogue; The Human microbiome project- HMP – which is hosted by the NIH and JGI’s extensive program and archive.
New Tools
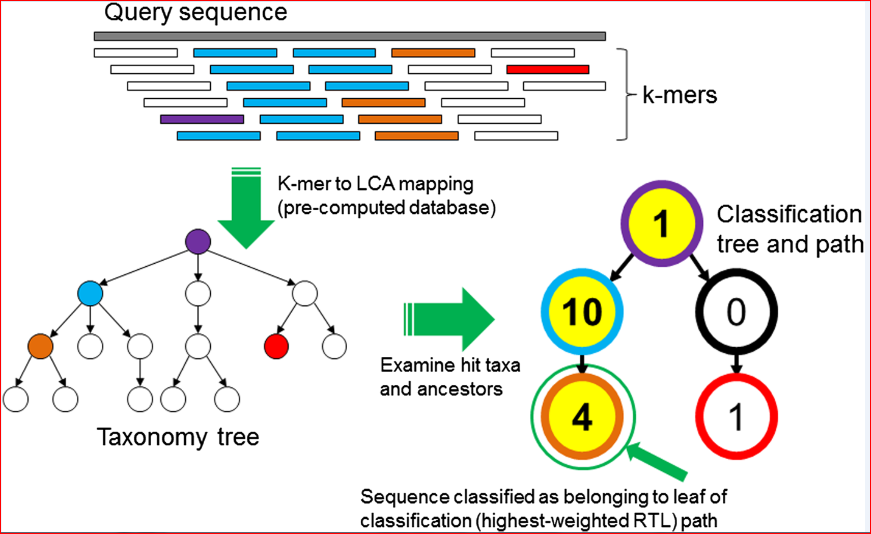
Wood and Salzberg recently published an article in Genome Biology describing new, improved software for sequence classification by using exact matches: Kraken. They describe ‘an ultrafast and highly accurate program‘, which shows a significant improvement in assigning taxonomic labels to metagenomic DNA sequences in contrast to previou,s specifically designed, programs that have tended to be slow. This taxonomic label assignment is very important as many metagenomic samples have a largely unknown content at the time of sequencing. The figure below reflects the sequence classification algorithm included in their article.
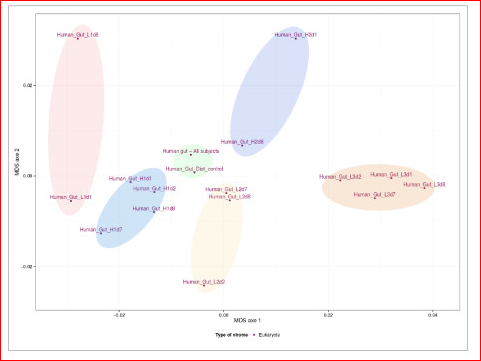
Although a fairly recent development in the genomics area, the description of pathogen or viral communities is becoming more appreciated as being of great importance to our knowledge of organism behaviour, disease and interactions. Published recently in BMC Bioinformatics, Simon Roux and colleagues from Clermont Université (France) have described new analysis tools for scientists working in this field. Their webserver Metavir 2: allows users to explore and analyze viromes (viral metagenomes) composed either of raw reads or assembled fragments through a set of adapted tools (Metavir is a description of the first version of the software). For example, this figure below shows a comparison of 16 unassembled human gut viromes and the assembled dataset based on their tetranucleotide compositions.
 New tools are being developed all the time in this fast-moving field, but how good are they at doing what you want them to do? Jorge Vázquez-Castellanos et al. recently published an article in BMC Genomics in which they compare the performance of a range of assemblers and taxonomic annotation software using simulated viral-metagenomic data for the analyses. They explain that the success of most assemblies is greatly hindered by the formation of chimeric contigs which are ‘ … formed at virtually any taxonomic level or function, regardless of the stringency of the parameters and the existence of reads of bacterial origin in the dataset.‘ The authors propose some specific tools for optimal analyses under differing contexts.
New tools are being developed all the time in this fast-moving field, but how good are they at doing what you want them to do? Jorge Vázquez-Castellanos et al. recently published an article in BMC Genomics in which they compare the performance of a range of assemblers and taxonomic annotation software using simulated viral-metagenomic data for the analyses. They explain that the success of most assemblies is greatly hindered by the formation of chimeric contigs which are ‘ … formed at virtually any taxonomic level or function, regardless of the stringency of the parameters and the existence of reads of bacterial origin in the dataset.‘ The authors propose some specific tools for optimal analyses under differing contexts.
New Applications
Microbial communities are known to be important to human health. The HMP project seeks to ‘Generate microbiome taxonomic, metagenomic and functional data from clinical biospecimens obtained from a cohort(s) of carefully-phenotyped subjects with a specific disease or health state. ‘ and ‘Combine the microbiome and host data to produce a community resource‘. These communities are also important in ecology, biofuel production, agriculture, and animal health. A recent short report in Virology Journal by Lu et al. entitled ‘Genomic variation in macrophage-cultured European porcine reproductive and respiratory syndrome virus Olot/91 revealed using ultra-deep next generation sequencing’ uses an ultra-deep sequencing methodology, without prior knowledge of the sequence, to construct the complete genome of a variant of an economically important livestock disease called PRRSV Olot/91. Describing how application of this methodology could be used to explore this disease further, the authors explain:
‘Rare variants that constitute minor fractions of the heterogeneous PRRSV population could successfully be detected to allow further exploration of microevolutionary events.’
New Challenges
Wommack and colleagues used mock viral communities to examine the influence of pooling on population-scale analyses. Multiple displacement amplification (MDA) has been one of the most commonly used methods for amplifying genomic DNA from the environment and is effective at amplifying minute amounts of DNA. Reported biases, however, include preferential amplification of single stranded circular DNA and non-uniform amplification of linear genomes. The authors found that in pooled and single reaction multiple displacement amplification treatments, ‘sequence coverage of viral populations was highly variable and coverage patterns across viral genomes were nearly identical, indicating that initial priming biases were reproducible and that pooling did not alleviate biases.’ However, control unamplified sequence libraries showed a relatively even coverage across phage genomes.
All these metagenomics examples are from publications in 2014. Looking back into 2013 provides us with an even more extensive variety of research on this subject, arising out of many varied biological contexts. Metagenomics remains an extraordinarily useful tool in furthering our understanding of (amongst others) human and animal health, ecology and economics. The articles below give a brief glimpse into the diversity of applications that this field is now investigating. We look forward to seeing what the rest of 2014 – and beyond – has in store.
Some other recent BioMed Central articles:
Coghlan ML et al.: Metabarcoding avian diets at airports: implications for birdstrike hazard management planning. Investigative Genetics 2013, 4:27. link
Ross, E.M. et al.: Metagenomics of rumen bacteriophage from thirteen lactating dairy cattle. BMC Microbiology 2013, 13:242.link
Berman HF and RileyLW: Identification of novel antimicrobial resistance genes from microbiota on retail spinach. BMC Microbiology 2013, 13:272. link

One Comment