New research published today in Clinical Epigenetics questions the methods used in some studies for assessing DNA methylation in cancer, calling for the use of only quantitative techniques. This is a guest blog by Dr Annette Lim (Peter MacCallum Cancer Center, Australia) and Dr Alexander Dobrovic (Ludwig Institute for Cancer Research and Olivia Newton-John Cancer Research Institute, Australia) to explain more about the importance of how we measure DNA methylation in cancer.
Given the critical role of methylation in embryogenesis, it is not difficult to expect that dysregulation of methylation will be a fundamental element in the evolution of uncontrolled cell cycling within the cancer genome. As such, the identification of altered methylation in tumours has held promise of unlocking key steps of carcinogenesis, with the “holy grail” of providing opportunity for therapeutic manipulation and utility as clinical biomarkers.
However, the method of assessment of methylation and the characteristics of the clinical samples examined are likely to influence findings, and the clinical interpretation of the results.
We demonstrated that the use of non-quantitative versus quantitative methodology altered the number of methylation events detected within a uniform cohort of clinical samples. Furthermore, cut-off criteria utilised to account for the sensitivity of the assays and to exclude low levels of methylation introduced from normal infiltrating cells, not surprisingly modified the total number of events detected.
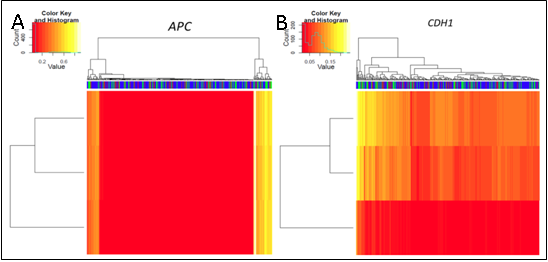
As such, the findings of our correlative analysis altered according to the different methodology used. Any correlative analysis will be influenced by the imputed data.
It is challenging to ponder what the important steps are in a study’s design that will minimise variability within the data that is introduced by methodology, given the already inherent heterogeneity that occurs within a tumour genome. In an idealistic scenario where the detection of a biomarker would be simple, for a uniform cohort of samples, a single clinically routine biopsy would provide DNA sufficient to represent the entire tumour’s biology for a single tissue histotype, the detected methylation would be homogeneous in both its quantity and distribution, and the methylation event would be highly penetrant.
However, this scenario is not common for many molecular events in tumourigenesis. The scenario assumes that we have already defined comprehensively the location and impact of all methylation events, and excludes the influence of other epigenetic/molecular events.
How then do we cope with the reality that humans, their tumours and methylation within tumours are in many instances only heterogeneous? Beyond what is already known, how do we progress from here in understanding whether methylation events in different cancers are therapeutically or biologically critical events? A key step in the future may include the ability to analyse methylation patterns at the single molecule level.
Though the answers to these questions require further research, these steps will include what we already know are crucial elements of study design; to recognise the characteristics of what we are examining and to consider the limitations of the methodology used, for the interpretation of correlative analyses.
As cancer continues to devastate lives around the world, an open-minded approach to research beyond binary interpretations, will hopefully reveal the answer. In the end, with the pervasive heterogeneity that we deal with in real life, it is possible that regardless of the methodology used that everyone will be correct about something some of the time!

- Raising funds for genetic diseases - 23rd September 2016
- The Epigenetics and Chromatin Clinic - 9th November 2015
- Resurrecting one of the oldest genetics journals - 23rd October 2015
Comments