Many of the most frightening human viruses have only recently made the jump from another species. These ‘zoonoses’ include the pandemic virus SARS-Cov-19 (which famously derived from a bat virus), and can be sourced from species both closely related to humanity (such as HIV originating from apes) and from species more distally related (such as avian flu from birds). By simmering in natural reservoirs and infecting Homo sapiens at unexpected times, zoonotic diseases present a formidable, shadowy foe for public health. Vector-borne viruses (known as the arboviruses) are one of many families that include zoonotic members. But this is a human-centric view (a way that we as a species are prone to thinking), as the trajectory of a virus’ host can also go the other way. A recent study has shown that not only are these anthroponotic (human-to-animal) jumps common, they may even occur more frequently than zoonotic paths.
A team of scientists in the UK from University College London and The Francis Crick Institute, Cedric Tan. Lucy van Dorp, and Francois Balloux, decided to use the plethora of viral genomic data already accumulated on public databases to investigate how viruses take the plunge into a new host animal. From the NCBI Virus resource they could scour almost 60,000 total genomes, each maintained to a threshold of quality (it is worth remembering that, as studies like this exemplify, public data enables science to be conducted without even getting up from your chair)! This search cast a wide net across the extent of viruses infecting vertebrates, incorporating 32 viral families and 62 host orders.
Although a taxonomy of viruses exists, the standards are not always maintained to the degree that they should be when reporting new viral sequence, and the lexicon itself is debated due to the dynamic nature of virus genomes. To avoid pre-defined family groups from obscuring the results, Cedric and co. decided to cluster the different viral genomes together based purely on similarity. This “species-agnostic” approach bypassed the taxonomy, although the grander viral family definitions were still used to ascertain where these clusters belong. As a sanity test, the researchers’ groupings corresponded strongly with those of the official taxonomy.
To track when the viruses made a host jump, the collated sequences were compared against one another, and the similarities and mismatches between the sequences used to produce a phylogeny – a prediction of the ‘family tree’ demonstrating these viruses evolved over time. Going back in time, at the point where two lineages converge into a common ancestor, one can estimate the most probable host species of said ancestor based on the hosts of viruses today. For example, if most of the lineages from a given ancestral node of the tree infect horses in the modern day, the most likely explanation is that the ancestral virus did too.
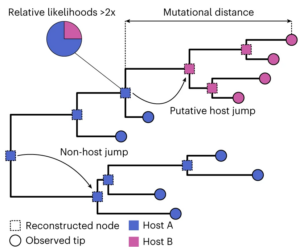
This strategy predicted nearly 3000 host jumps among the viral sequences studied, a fifth of which involved humans as either the original or the new host. The ample enrichment of Homo sapiens in this list is not surprising given how often humans would be sampled for viral sequence, and hence contribute to the data. What was more surprising, however, was what the team found when they compared the directionality of these predicted host jumps – nearly double the number of transitions were anthroponotic, as opposed to zoonotic. This strongly suggests that, although animal-to-human jumps are the most studied, human-to-animal jumps are actually more prevalent.

It can be easy to forget that one of the many threats that humans pose to animals is giving them disease. The authors highlight that man-sourced outbreaks among other species have been observed before to devastating effect, such as multiple deadly respiratory virus outbreaks among chimpanzees in Tanzania. Many of the predicted anthroponotic leaps are associated with the coronaviruses and influenza viral clusters, which makes sense given the number of spillover events observed for SARS-CoV-2 into other animals, such as cats. Across the 21 viral families that the author’s report, the higher rate of anthroponotic jumps is observed in sixteen of them, making this pattern generalisable across viruses.
To further understand these transitions, the researchers investigated how mutational rate is linked to a new tropism. As may be expected, a host jump was associated with higher average mutational changes to the viral genomes, suggesting that either more mutations begets a host jump or a host jump selects for mutants. The number of mutations needed to make the leap is not the same for all viral groups. A virus family which already exhibits a broad host range, on average, requires fewer additional changes to add a new tropism to their roster.
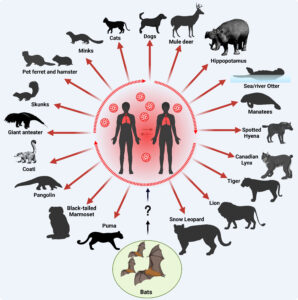
In this study the authors did not focus much on arboviruses, although a number of families from this clade were part of the dataset. Perhaps here there is an opportunity for another plucky scientist to make important discoveries from digging into the information? Indeed, vector-borne diseases have a particularly intimate relationship with host jumps as it is a natural part of their life cycle, and by definition they infect a minimum of two different species (species with extremely low similarity at that). A quick look at the host transitions detected for the Togaviridae (the viral family that includes chikungunya virus) and the Flaviviridae (which includes dengue and Japanese encephalitis viruses) mostly highlights the diversity of the predicted sources and sinks of human viral jumps – with marmosets, monkeys, horses, pikas, pigs, and many species of bird featuring. That isn’t to say that all these species can transmit deadly disease, there may be transitory species in-between the source host and the new one detected, but it does highlight the great world of viral diversity that could birth an emerging human pathogen.
While the world watches for the next zoonose to wreak havoc on global stability and claim innumerous lives, the animals threatened by the reverse zoonotic pathway have been cast to the sidelines. This study shows that the animal kingdom may have more to fear from human viruses than the other way round, and could prompt a rethink of our place in the natural world.

_(7068198095).jpg){kind=link}
Comments