The mouse bile-duct tapeworm Hymenolepis microstoma (photo credit: PD Olson).
Tapeworms are ubiquitous parasites of all classes of vertebrates and have complex life cycles that typically involve at least one invertebrate intermediate host and one vertebrate final host in which the adult, segmented worm resides in the intestinal system. Human infection is most severe when it involves us playing the role of the intermediate host, acquiring larval forms of the tapeworm which locate outside of the enteric system, often in association with the central nervous system. For example, human infection with the larvae of the ‘pork’ tapeworm Taenia solium is estimated to be responsible for a third of cases of epilepsy in Latin America.
Although such species are important to investigate, their life cycles cannot be practically maintained in the laboratory and hence much of our fundamental understanding of tapeworm biology is based instead on species with life cycles involving beetles and rodents, as such hosts are themselves used as laboratory models.
Characterisation of the genomes of parasitic worms led by the Parasite Genomics Group at the Sanger Institute represents one the most important global advances in our efforts to conquer the chronic diseases caused by these pathogens. In roughly a decade, the genomes of the most important species of flatworm (i.e. platyhelminths) and roundworm (i.e. nematodes) parasites have been characterised and the data made freely available to all.
Some of these genomes have now been assembled to the level of complete chromosomes, allowing investigation not only of genome content but also of the genetic landscape arrayed along their chromosomes. One of these is the mouse bile-duct tapeworm, Hymenolepis microstoma, an important laboratory model for which a draft genome was published in 2013.
Getting it together
The first genome level sequencing technologies were based on a divide and conquer approach: the genome is fragmented into millions of short (100s of bases) pieces which are then sequenced in parallel, generating millions of short ‘reads’ that must be assembled on a computer. While these technologies are sufficient in terms of covering all or most of the bases, short reads are problematic to assemble, as repetitive and low complexity sequences in the genome mean that many reads cannot be unambiguously aligned to single positions.
As a result, most characterised genomes to date are still made up of far more un-assembled fragments of sequence than the number of chromosomes the organism has, obscuring their syntenic relationships (i.e. the relative positions of the various genetic elements).
More recent technologies allow for sequencing of long reads – very long reads – generating contiguous sequences of hundreds of thousands to millions of bases, while complimentary approaches such as optical mapping (a non-sequence based approach to physically mapping the relative positions of chromosome fragments) provides additional evidence to aid in higher-level assembly. These technologies were used to transform the draft genome of H. microstoma into a fully assembled, chromosome-level reference genome – the first entirely resolved genome of a representative of the Lophotrochozoa: the great animal group encompassing molluscs, annelids, flatworms and a diverse array of smaller phyla of invertebrate animals.
Junk most important
A fully characterised and assembled genome is invaluable in research for many reasons, not least because it is free from sampling error (e.g. is a gene really missing, or has the genome been incompletely characterised?). Beyond the content of the genome, it also provides the opportunity to investigate its architecture: how the different elements of the genome – from the parts that code for proteins to those that represent genomic invaders – are arrayed along the different chromosomes (the longest contiguous stretches of a eukaryotic genome).
It has been known since the early days of sequencing of the human genome that much of it is comprised of short, non-coding sequence motifs. Initially known as ‘junk DNA’, their importance to genome evolution is still only starting to be appreciated. These sequences are the result of ‘transposable elements’ (TEs) which are bits of foreign, viral DNA that are incorporated into the genome and are variously removed or amplified in copy number through the course of evolution. All eukaryotic genomes investigated to date contain TEs, which can comprise over half of the genome in some species.
Today it is accepted that far from being junk, TEs are in fact responsible for some of the most significant aspects of genome evolution, such as gene duplication and reshuffling. But they are also responsible for the evolution of linear chromosomes themselves (a hallmark of eukaryotes) which are ‘capped’ (terminate) by short sequence motifs called telomeres. These 6 base sequence repeats act to maintain both the linearity and full lengths of chromosomes during replication.
Meanwhile, a much longer (~370 base) sequence motif called the centromere acts as the site of spindle attachment, enabling homologous chromosomes to segregate during cell division. Centromeres too have their evolutionary origins in TEs, which paradoxically have species-specific sequence identities driven by the dynamic evolution of TEs, despite performing an entirely conserved and fundamental role in mitosis.
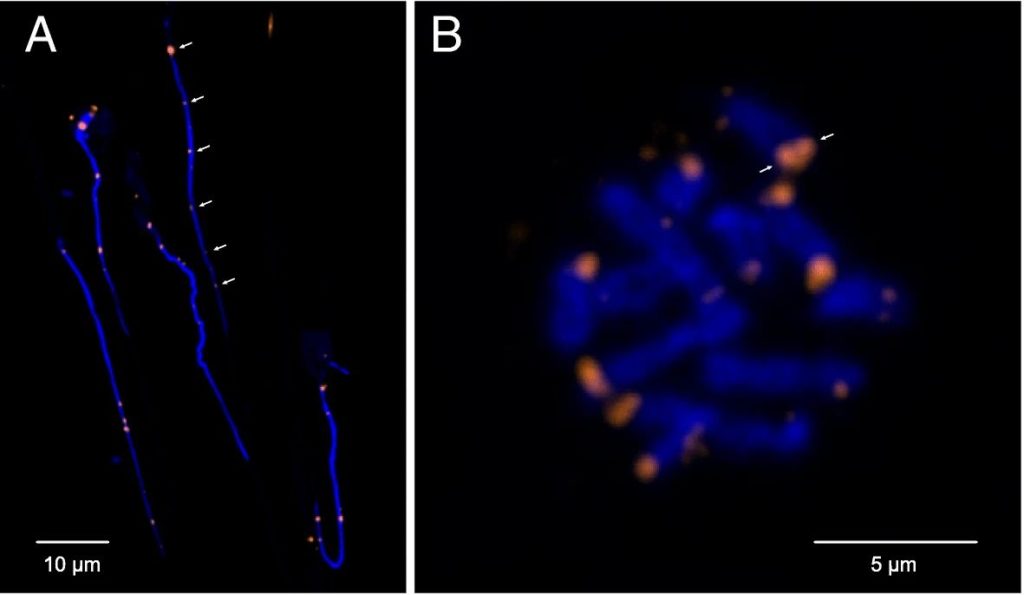
Losing your telomeres
Complete assembly of the H. microstoma genome revealed that its chromosomes are capped by telomeres only on one end, whereas the opposing ends terminate instead with what turned out to be centromeric sequence arrays. Classically, the position of the centromere relative to the ends of chromosomes has been used as a means of describing a species ‘karyotype’. Those found to be near the ends of the chromosome are known as ‘telocentric’ (‘near the telomeres’) and, given the limited resolving abilities of karyological techniques, were assumed to nevertheless terminate in telomere sequence.
However, the H. microstoma genome definitively shows that chromosomes can indeed terminate in centromeric arrays, which presumably came to replace the telomeres through the course of evolution.
Being terminal necessitates that the centromere plays the role of a telomere in protecting chromosome ends. At the same time it must also maintain its ancestral role as a substrate for spindle attachment during cell division. However, evolving to play a dual role is less than straight forward, as there are telomere-specific proteins that interact directly with telomeric sequences to maintain chromosome length homeostasis, and these presumably must also interact with the centromere sequence motif in the case of H. microstoma. This and other implications with respect to the underlying mechanisms that orchestrate these fundamental processes in H. microstoma require further investigation. Meanwhile, whether or not terminal centromeres are found in other species described with telocentric karyotypes awaits the full assembly of additional genomes.
Parasites are just organisms
It is common conceit even among biologists to consider parasites as if they were wholly separate from free-living animals, having nothing to teach us about (ultimately) our own biology. In the same vain, any novelty in their biology is typically linked to their parasitic lifestyles. But parasitism is simply a trophic strategy that has been exploited by at least some lineages within the majority of major organismal groups; and few would argue, for example, that we have nothing to learn from the study of herbivores than understanding herbivory. These findings in a tapeworm point to fundamental lessons in chromosome evolution applicable to all organisms – not to the affects of ‘parasitism’ on the genome.

Comments