The bottleneck of tumor genome interpretation
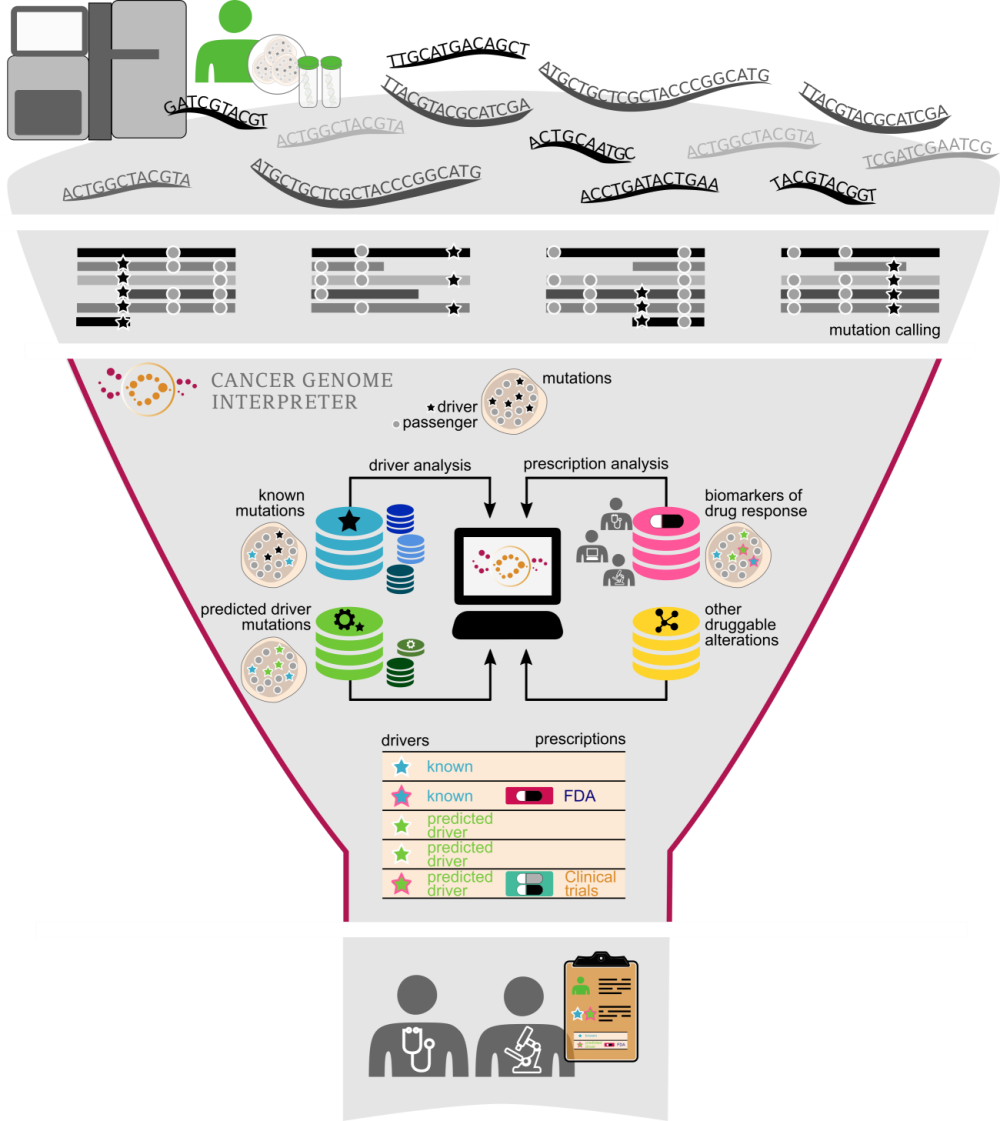
Hundreds of tumor genomes are sequenced routinely around the world every day as part of research projects or to guide therapeutic decisions. In some cases only a small portion of the genome—the part that is most likely to inform treatment options—is sequenced, while in others the coding region of the genes, or even the whole genome, is sequenced.
In both clinical and research applications, the DNA sequencing step is followed by calling the somatic variants in a tumor genome. However, the major bottleneck of tumor analysis is currently the identification of those variants that have biological or clinical relevance. This is because most of the alterations observed in a tumor, including those in well-known cancer genes, are of uncertain significance. Moreover, although new highly potent and selective anti-cancer drugs have been developed and tested in recent years, information on the tumor variants that influence their response is patchy across the literature and in several specialized resources.
To address this challenge, we have developed the Cancer Genome Interpreter, an open platform that carries out extensive annotation of the tumor variants to guide clinicians and researchers in their prioritization.
Learning from cancer cohorts to interpret the genome of individual tumors
The interpretation of cancer variants is based on knowledge acquired from the analysis of cancer genomics and the accumulation of results of trials on the effectiveness of drugs on cohorts of patients or individual cases.
Decades of research on cancer genetics and the more recent mining of large tumor cohorts, sequenced by large consortia like TCGA and ICGC, have led to the identification of the genes that carry cancer-causing mutations (i.e., the drivers of cancer). As a result, stable catalogs of experimentally validated (Cancer Gene Census) or bioinformatically identified (such as IntOGen, developed in our group) cancer genes have emerged.
However, not all mutations in these cancer genes are drivers. In fact, almost 9 out of 10 of the mutations found in cancer genes are variants of unknown significance. This observation highlights the importance of tools able to further assess their oncogenic relevance.
By studying mutational patterns across thousands of tumors and healthy samples, we can distinguish which of the mutations affecting these genes are most likely to be tumorigenic.
By studying mutational patterns across thousands of tumors and healthy samples, we can distinguish which of the mutations affecting these genes are most likely to be tumorigenic. For example, driver gain-of-function missense mutations tend to cluster in specific regions of oncoproteins. These clusters become apparent when thousands of tumor genomes are analyzed together. We are also able to identify driver genes that tend to bear truncating mutations, i.e. tumor suppressor genes. And in both cases, mutations that occur in protein domains depleted of germline variants in healthy samples are more likely to be pathogenic.
All accumulated knowledge about oncogenic mutations and the information gained from the analysis of large sequenced cohorts are used by the Cancer Genome Interpreter to identify those mutations that are most likely to be drivers in a newly sequenced tumor.
In parallel, specific inhibitors of oncoproteins are tested in clinical trials—in some cases coupled with tumor DNA sequencing—to determine their effectiveness and safety in cohorts of patients. These studies have identified genomic biomarkers of drug response, which we gathered in a database organized by their level of clinical relevance. The Cancer Genome Interpreter matches this information with the variants found in the newly sequenced tumor, taking into account the interactions of co-occurring variants that may affect drug response.
Several initiatives from individual institutions have recently started to aggregate these biomarkers into various resources and to organize them to meet the needs of distinct users.
However, the task is challenging due to the amount of information continuously being generated and the necessity to classify biomarkers based on their supported evidence. To pool the efforts of these institutions (including our own database of biomarkers) and unify their curation and annotation standards, we recently launched the Variant Interpretation for Cancer Consortium.
The Cancer Genome Interpreter is an important milestone toward the goal of efficient cancer genome interpretation in clinical and research oncology settings. The refinement of the annotation of tumorigenic mutations on the basis of the analysis of recently sequenced genomes, and the incorporation of newly discovered molecular features of the tumors that inform their treatment, will further improve the usefulness of the tool as our knowledge evolves.
Comments