In this guest post, Dr Andrew Teschendorff of University College London and the CAS-MPG Partner Institute for Computational Biology, Shanghai, examines a new Genome Medicine study.
In an exciting research article published today in Genome Medicine, Rafa Irizzary and colleagues provide evidence for a gradual systems-level deregulation of the epigenome in stages prior to the onset of cancer and which later is seen to progress further in cancer. Thus, these insights could potentially lead to a clinical test with the ability to predict cancer risk in cells that are not yet malignant.
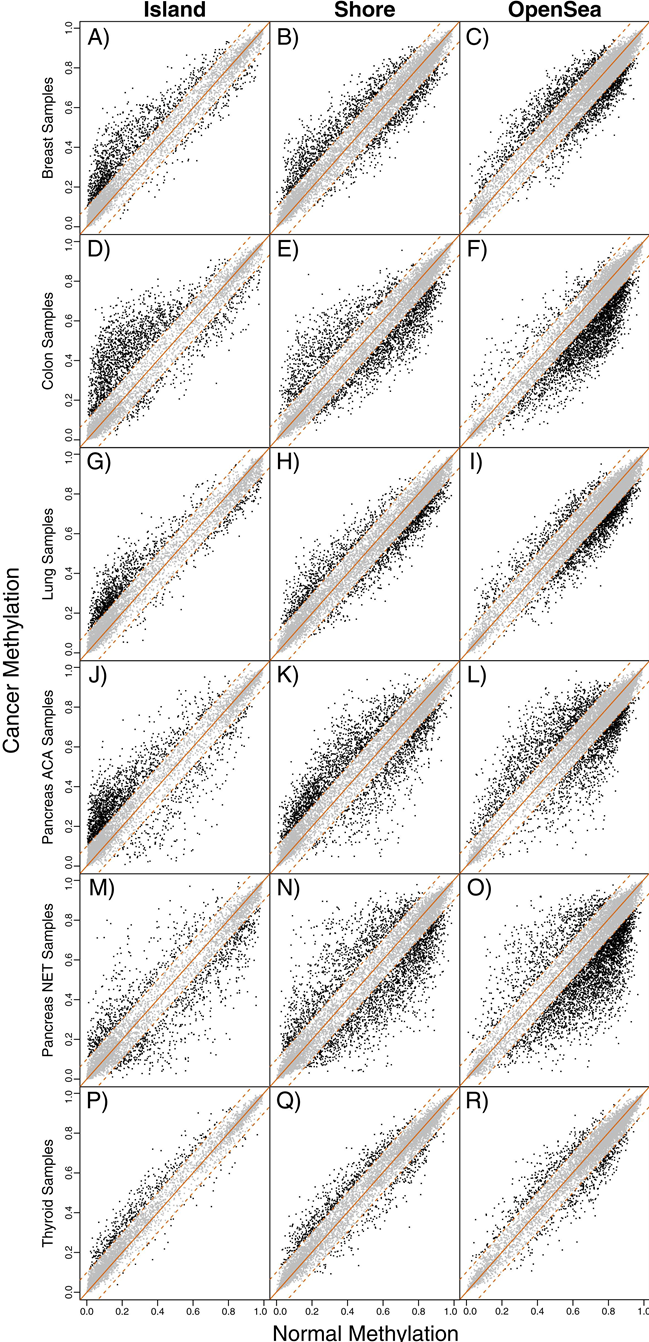
The authors focused on a specific epigenetic mark, known as DNA methylation, a molecular modification of DNA which can regulate the activity of nearby genes. They developed a novel statistical algorithm and applied it to a large set of DNA samples from different tissue types and disease stages, including cancer.
By doing this they were able to demonstrate that DNA methylation changes are progressive during carcinogenesis. What’s more, they showed that most of these changes occur in large spatially organized genomic regions of DNA, involving loss of DNA methylation, called “hypomethylated blocks”.
Many of these blocks coincide with genomic regions that are normally attached to the cell’s nuclear membrane, with genes in these regions normally switched off. Hypomethylation of these blocks may lead to genes located within these blocks being wrongly re-activated, and more generally to an increase in the variability of gene activity patterns. The authors hypothesize that these may increase cellular heterogeneity and so underpin cancer development.
Irizarry and colleagues’s work further demonstrates that the relatively infrequent increased DNA methylation observed in cancer occurs in smaller, localized, regions immersed within these large hypomethylated blocks. This unifies two long-standing observations about cancer DNA methylation patterns.
Most importantly, this unification of what previously appeared as two distinct epigenetic phenomena, points towards a common systems-level deregulation of the epigenome, which may in future serve as an important risk marker for carcinogenesis.
Indeed, earlier work, also published in Genome Medicine, demonstrated that increased DNA methylation occurring locally, at specific genes with roles in normal development, could be used to predict the risk of neoplastic transformation (the conversion of normal tissue into a malignant tumor) three years in advance of such transformation taking place.
By adopting a more recent and comprehensive technology, Irizzary and colleagues were able to measure DNA methylation, not only near genes, but also in the large genomic regions (denoted as “open seas”) between genes, demonstrating that the previously observed DNA methylation increases is a local phenomenon subsumed by the larger hypomethylated blocks.
It remains unclear what drives this systems-level deregulation of the epigenome, and in particular which gene activity changes are important for cancer development. It is possible that deregulation of genes, which modulate and control the epigenome, cause the observed systems-level deregulation of the normal DNA methylation landscape.
This modified DNA methylation landscape could then lead to de-repression of genes that cause the development of tumors, as well as to a simultaneous repression of genes that promote differentiation and normal homeostasis.
As shown in the context of the cervix, normal cervical cells with such an aberrant epigenome may be at an increased risk of becoming cancerous. Prospective studies, which can access the normal cells of origin of other solid tissue cancers and measure their DNA methylation patterns years in advance of cancer diagnosis, will be needed to address these important questions. Clinical application will, however, most likely require the use of easily accessible surrogate tissues (eg. blood, buccal or cervical smears), which may record epigenetic fingerprints associated with cancer risk factors and which can be accessed routinely and non-invasively.
Another key question raised by the current study, and which will be important to address in in the context of somatically acquired (i.e. non-heritable) cancers, concerns the relative order in which the epigenetic and genetic changes take place. For instance, is the deregulation of the DNA methylome in disease stages prior to cancer driven by genetic or epigenetic mutations?
Comments