 Rare diseases are notoriously difficult to diagnose and treat. There are a number of reasons for this, but chief among them is the very fact of their rarity. With only small patient numbers, it can be challenging to carry out research leading to new diagnostics and treatments.
Rare diseases are notoriously difficult to diagnose and treat. There are a number of reasons for this, but chief among them is the very fact of their rarity. With only small patient numbers, it can be challenging to carry out research leading to new diagnostics and treatments.
Today is Rare Disease Day, a movement which aims to bring together everyone involved in trying to change this state of affairs, from practitioners to policymakers, social services to scientists, policy makers, and, crucially, patients. So it seems appropriate for us to highlight some promising new research into Apert syndrome, published today in BMC Developmental Biology.
Apert syndrome is a rare congenital disorder where the skull and facial bones fuse prematurely, a symptom known as craniosynostosis, alongside malformations of the hands and feet. The malformations of the skull seen in patients start prenatally and continue to develop and worsen following birth. There is no preventative treatment and children usually have to undergo surgery, in some cases multiple rounds, to separate the fused bone.
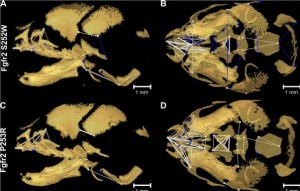
The syndrome is caused by gain of function mutations in fibroblast growth factor receptor 2 (FGFR2), which plays a role in various stages of embryonic development and especially bone development. The majority of Apert syndrome cases are due to two specific mutations in FGFR2, Ser252Trp or Pro253Arg, which in turn cause slight variations in the manifestation of the disease.
Apert syndrome is very difficult to study in humans, but mouse models of the most common mutations do exist. The new research by Professor Joan T Richtsmeierand colleagues demonstrates that mice with the condition show a consistent pre-natal growth pattern which, if replicated in humans, may help us to explore methods for earlier diagnosis and improve treatment options.
Micro-computed tomography was used to build up 3D images of the mice skulls. One of the great difficulties with these studies is the ability to accurately control between differences seen due to individual development patterns and the mutations themselves. Prof Richtsmeier explains the statistical tests developed to address this issue in an accompanying Q&A in Biome.
The data from these 3D images allowed the pattern of the developmental change to be mapped and revealed the consistency in the growth patterns of the skulls of the mutant mice, as well as key differences between the two FGFR2 mutations.
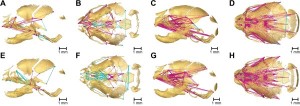
This has implications for Apert syndrome patients as the ability to predict how the skull will continue to develop could lead to more tailored surgery, including the ability to take future skull development into account. It would also allow doctors to provide more information to both parents and children about the realistic outcomes of such treatments.
Philippa Harris
Latest posts by Philippa Harris (see all)
- The Tef genome: deciphering one of Ethiopia’s key crops - 14th August 2014
- Why I study malaria: Dr Francis Ndungu on research, working in Kenya and open access - 25th April 2014
- New research sheds light on the development of a rare disease - 28th February 2014
Comments