A recent paper in BMC Microbiology argues that delineation of bacterial species based purely on whole genome sequences is both feasible and desirable. In this guest blog, the authors outline why.
In the early eighteenth century, in his Systema Naturae, Linnaeus provided the first workable hierarchical classification of species, based on the clustering of organisms according to their phenotypic characteristics. Over a century later in On the Origin of Species, Darwin added phylogeny, i.e. the history of organismal lineages over time, to biological taxonomy, while also emphasizing the arbitrary nature of biological species. Despite concerns over the capricious nature of taxonomic boundaries, the pragmatic reality and utility of the species concept continues to inform the theory and practice of microbiology and a stable species nomenclature underpins the diagnosis and monitoring of pathogenic microorganisms.
For several decades, when defining a bacterial species, microbial taxonomists have used a multi-stranded approach that relies on morphological, physiological and biochemical information. These in turn rely on growth, usually in pure culture, in the laboratory, which may not be achievable for many bacterial species. This polyphasic approach also relies on techniques, such as DNA-DNA hybridization, that are time-consuming and difficult, particularly when compared to the ease of modern genome sequencing. Motivated by recent advances in high-throughput sequencing, we, along with other researchers, have begun to explore the suitability of genome sequence data for the purposes of bacterial species delineation and taxonomy.
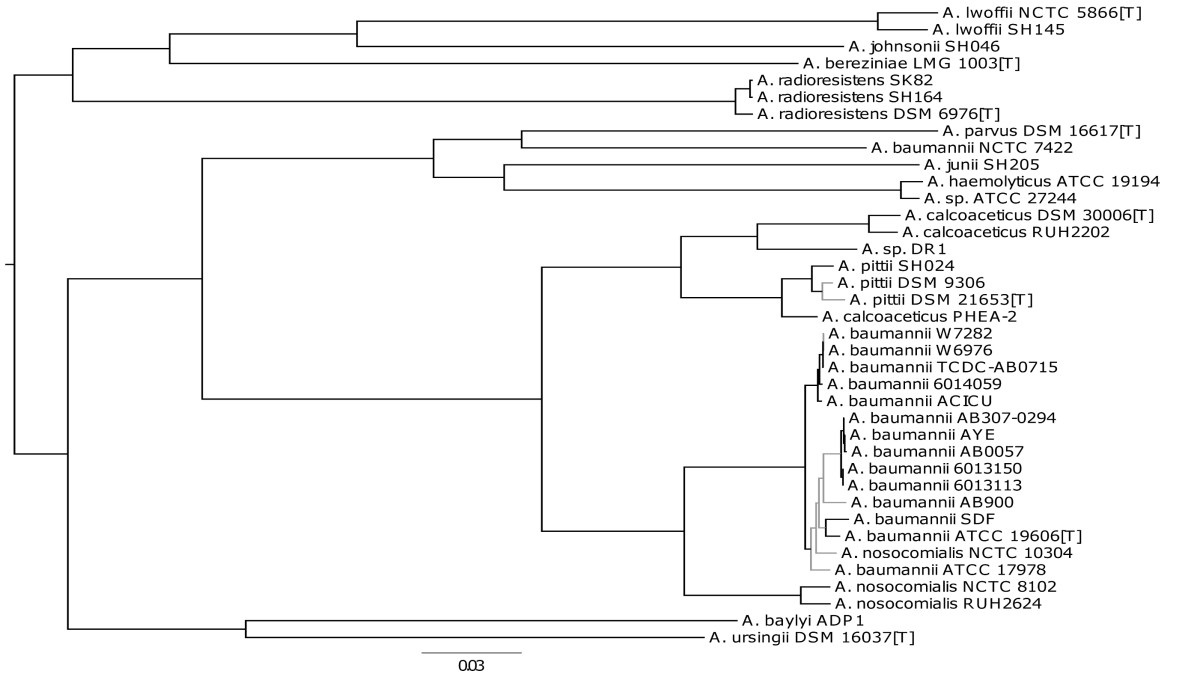 In our recent BMC Microbiology publication we explore, using the genus Acinetobacter, whether comparative analysis of genome sequences could rival classical DNA-DNA hybridization and phenotypic approaches. To date there are 27 species of Acinetobacter described including the emerging pathogen Acinetobacter baumannii. However, A. baumannii is highly related to three other Acinetobacter species (A. calcoaceticus, A. pittii and A. nosocomialis) and it can be difficult to distinguish the four based on phenotype alone, making the genus an ideal test case.
In our recent BMC Microbiology publication we explore, using the genus Acinetobacter, whether comparative analysis of genome sequences could rival classical DNA-DNA hybridization and phenotypic approaches. To date there are 27 species of Acinetobacter described including the emerging pathogen Acinetobacter baumannii. However, A. baumannii is highly related to three other Acinetobacter species (A. calcoaceticus, A. pittii and A. nosocomialis) and it can be difficult to distinguish the four based on phenotype alone, making the genus an ideal test case.
We found that a combination of a phylogenetic analysis of shared genomic regions and calculations of average nucleotide identity across genomes provided a simple yet informative and powerful method suitable for bacterial species delineation. Our results are consistent with the currently accepted taxonomy of genus Acinetobacter, while also identifying three misclassifications of strains in collections or databases. The methodology that we propose provides a scalable and uniform approach that works for both culturable and non-culturable species. It is faster and cheaper than traditional taxonomic methods; it is easily replicable and transferable among research institutions, providing a scalable, uniform and consistent framework for taxonomic classification; and lastly, it falls in line with Darwin’s vision of “classification becoming, as far as is possible, genealogical”.
Are there any drawbacks to this approach? Well, it does mean turning one’s back on more than a century of history: for many bacteriologists, it seems intuitively wrong to ignore phenotypes completely. After all, this is a conservative discipline that has allowed Shigella to stand as a genus name, when phylogenomics tells us this is just yet another pathovar of E. coli. More importantly, sequencing approaches are in a constant state of flux, driven by relentless technical innovation, with no stable standardised approach to producing a draft or complete genome foreseeable in the near future. This presents problems for a discipline wedded to “standard operating procedures”.
Yet the direction of travel is clear. We are hurtling towards a technological singularity in sequencing, when sequencing becomes so quick, cheap and easy that it will become the preferred technology for many applications in microbiology. When sequencing platforms arrive that deliver long read lengths and high accuracy, metagenomic sequencing of clinical samples will also be widely adopted. The challenge ahead is to adjust the theory and practice of bacterial taxonomy and clinical diagnostic microbiology to cope with the coming revolution. The times they are a-changing!
Jacqueline Chan, Mihail Halachev and Mark Pallen
University of Birmingham

Comments