There is as yet no cure for any neurodegenerative disease, and although we know there are common themes – protein misfolding, mitochondrial dysfunction, microtubule impairment – how these contribute to disease remains elusive.
Research into this question was one theme permeating the 46th annual meeting of the Society for Neuroscience this year, with a range of symposia showcasing not only what we’ve learned but the tools that have made new insights possible. Alzheimer’s disease, the commonest of the dementias, was a particularly prominent topic.
Model mice
Central to the tools for research into neurodegenerative disease are mouse models that recapitulate their progress. But these are not easily achieved.
Most mouse models of Alzheimer’s disease, for instance, have until recently been based on over-expression of human genes implicated in its pathology – in particular amyloid precursor protein (APP). APP gets cleaved into several fragments; one, Aβ (specifically the Aβ42 form) forms the plaques characteristic of Alzheimer’s disease, and is thought to be a crucial mediator of disease progression.
But onset of symptoms is early in these models – convenient for researchers, but not characteristic of the human disease, from which they also differ in other ways, and which are subject to the criticism that overexpression at a level unknown in the human disease may trigger stress responses, particularly as not only Aβ, but the other APP fragments (the biological function of which are unclear) are also being over-expressed.
Central to the tools for research into neurodegenerative disease are mouse models that recapitulate their progress
To address this, Takaomi Saido and colleagues created a new model which they described in a paper published in 2014 – an “APP knock-in” in which rather than overexpress a new gene, they introduced mutations associated with Alzheimer’s disease into the endogenous APP gene. This conferred typical hallmarks of the disease, including age-associated memory impairment, neuroinflammation, and Aβ accumulation / aggregation, resulting in plaque deposition, at physiological levels of gene expression.
These mice have been freely distributed to others researching Alzheimer’s disease, and two years on (symptoms develop over a period from 6 to 18 months), an entire session at this year’s Society for Neuroscience meeting was dedicated to follow-up investigations on the APP knock-in mouse.
Ilya Bezprozvanny (UT Southwestern) spoke on his recent work in APP knock-in mice investigating the role of calcium dysreglation in synaptic spine loss in Alzheimer’s disease. There is evidence from previous work that memory failure in Alzheimer’s disease is caused by loss of synaptic spines – specifically a class known as mushroom spines – in hippocampal neurons. Mushroom spine stability relies upon the activity of synaptic Ca2+/calmodulin kinase II (CamKII) , and thus on appropriate Ca2+ levels, which in turn depend on Ca2+ entry into the neural cells through the neuronal store-operated Ca2+ entry (nSOC) channel, which is regulated by stromal interaction molecule 2’s (STIM2).
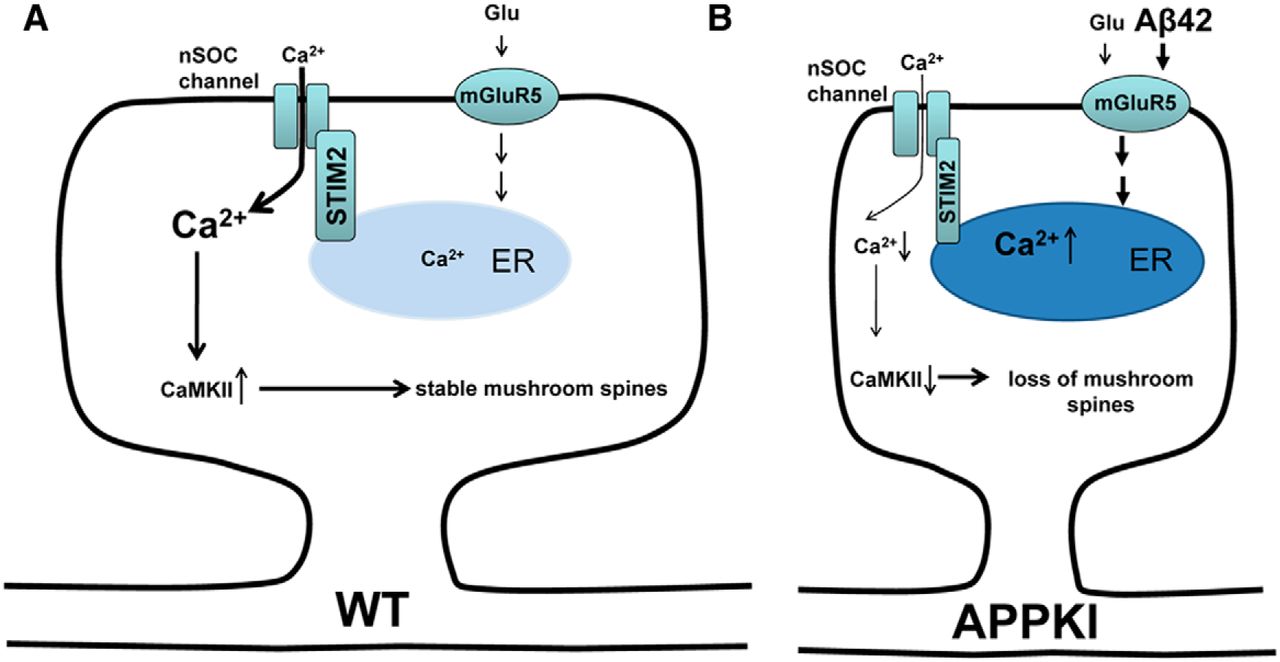
To this picture, Bezprozvanny and colleagues have now added an effect of Aβ42 on the mGluR5 receptor in cultured neurones from APP knock-in mice. Accumulation of Aβ42 leads to over-activation of the mGluR5 receptor, leading in turn to elevated Ca2+ levels in the endoplasmic reticulum and thereby down-regulation of STIM2. This in turn leads to reduced influx through nSOC and reduced activation of CamKII, culminating in mushroom spine loss in the hippocampal neurons.
Supporting this mechanism, they found that inhibition of mGluR5, or over-expression of STIM2, rescued mushroom spine loss in the APP knock-in neurons. Along with reports that STIM2 is down-regulated in sporadic Alzheimer’s disease, this work provides support for the “calcium hypothesis of neurodegeneration”, and suggests that nSOC might provide a therapeutic target for Alzheimer’s disease.
Limitations and combinations
Given the difficulty of assigning a role for any specific molecular mechanism in the development of dementia in Alzheimer’s disease, supporting evidence from human brain samples and from different mouse models is important to the case for any given mechanism.
Amantha Thathiah, (Pittsburgh Institute of Neurodegeneration) spoke on converging evidence she and colleagues collected in an international collaboration in 2015 for another potential therapeutic target – GPR3. In their study, two APP knock-in mice were used with two other mouse models to show that GPR3 expression levels is correlated with Aβ levels in all of the four models, with deletion of GPR3 leading to a reduction in plaque burden and alleviation of cognitive deficits.
They also reported that GPR3 is elevated in Alzheimer’s disease patients’ post-mortem brain tissue, providing further support for a role in Alzheimer’s disease pathology.
Supporting evidence from human brain samples and from different mouse models is important to the case for any given mechanism
One of the deficiencies of the APP knock-in mice is their mild, late-developing phenotypes, which make it difficult to identify behavioural deficits – hence the need for the two other mouse models to check this phenotype in relation to GPR3 expression.
Another deficiency of the APP knock-ins (also true of overexpression models) is their failure to show the neurofibrillary tau tangles characteristic of Alzheimer’s disease, or neuronal cell death.
These may in part reflect the limitations of working with mouse rather than human systems, and in an attempt to explore this possibility, Bart De Strooper VIB (/KU Leuven) implanted human induced pluripotent stem cells into new-born APP knock-in mouse brain to see how human neurons are affected by the Alzheimer’s disease pathology modelled in these mice.
These neurons also fail to develop the neurofibrillary tau tangles characteristic of Alzheimer’s disease, but they do contain the Aβ plaques (though at this stage it is unclear whether these plaques are donated by the mouse or generated by the human cells, following an initial triggering effect), and they undergo massive necrotic cell death.
This suggests that human neurons may be particularly vulnerable to Alzheimer’s disease pathology, and these chimaeras provide a means to start asking why that may be.
Tau tangles
Understanding how tau aggregates in vivo, and how aggregation relates to neurotoxicity, is a key question – not just in Alzheimer’s disease, but for a variety of neurodegenerative diseases
In an attempt to address the missing tangles problem, Takaomi Saido, with Takashi Saito, who spoke of it at the conference, has developed a mouse model with all six forms of human tau knocked in (there are only three forms in mice). They again have offered their mice to anyone interested in using them in their research.
In these mice, tau localises normally, and the mice appear to be healthy. However, when they are crossed with the APP knock-in mice, the tau is hyperphosphorylated and neuronal death is apparent – essentially, the “humanised” tau leads to an accentuation of disease phenotype (supporting the idea that Aβ is upstream of tau). Curiously, however, there were still no tangles present. Saito speculated that this might be because smaller aggregates of tau actually confer the pathogenicity.
Understanding how tau aggregates in vivo, and how aggregation relates to neurotoxicity, is a key question – not just in Alzheimer’s disease, but for a variety of neurodegenerative diseases characterised by tau aggregation, tauopathies, such as frontotemporal dementia.
With a view to this, Sarah DeVos, from Bradley Hyman’s lab in Massachusetts General Hospital, presented a tau sensor, which would provide a means to monitor tau aggregation. They use a FRET based approach, with a single vector containing both the repeat domain of tau (tauRD) fused with CFP and tauRD fused with YFP, to ensure transfection of both to a single neuron, following injection into the hippocampus.
DeVos reports that the FRET2 tau normally remains diffuse in the cell, though with the added benefit that spontaneous aggregation can be observed in both mutant tauopathy primary neurons and, with the aid of a cranial window, in a living mutant tauopathy mouse brain. This makes it possible to see what happens to a single neuron over-time after it develops an aggregate of tau, as well as carry out gene expression phenotyping to help determine some of the molecular consequences of tau aggregation in living neurons.
Diversity causes, diversity of approaches
As freely acknowledged at the meeting, no one model could be expected to replicate faithfully the disease phenotype. Most neurodegenerative diseases are heterogeneous, loss-of-function needs to be distinguished from gain-of-function mutations, and – most fundamental of all – we don’t yet know which hallmarks of disease reflect contributions to disease progression, and which are simply correlative. Untangling the mechanisms of neurodegenerative disease seems likely to take all the tools we can get.
Christian Matheou
Latest posts by Christian Matheou (see all)
- Mouse models, tau tangles and neurodegeneration in Alzheimer’s disease - 3rd January 2017
- Rooted in a changing environment - 23rd September 2016
- Learning from plants: new routes to engineering disease resistance - 22nd February 2016
Comments