
A study published today in BMC Biology has found that many published microbiome studies may have been contaminated. In this guest post, Susannah Salter and Alan Walker, authors on the paper, tell us more about what they found.
The last decade has seen amazing developments in DNA sequencing technology. One area that has benefitted tremendously from these advances is the field of microbiology, as it is now possible to characterise microbial communities (“microbiota”) at previously unimaginable depths.
As a result microbiota research is currently booming, led by many recent large-scale, world-wide initiatives such as The Human Microbiome Project, MetaHIT, and the Earth Microbiome Project, which use the power of sequencing to try and understand how microbial communities impact their surrounding environment.
Human-associated microbial communities, such as those present in the gut, oral cavity and vaginal tract, are of particular interest, since it is known that our microbial partners can impact health in many different ways. Sequence-based microbiota profiling has therefore been applied to a whole range of host-associated habitats, with the hope that these sensitive techniques can identify microbial agents associated with disease.
This includes low-biomass microbial populations from normally “sterile sites” in the body (such as blood or cerebrospinal fluid). These sites are interesting precisely because there is so little, if anything, living in them, so sparse cells may be undetectable by other means.
Contamination
One potential issue with the sensitivity of the new sequencing techniques is that it means that trace levels of contaminant DNA that could be introduced during sample handling are much more easily observed as well, and these may be difficult to distinguish from the “real” bacteria in samples. For samples that contain few bacteria (for example, blood samples, skin swabs, lung lavages) contaminating DNA could potentially drown out the DNA from the actual microbes that we are interested in.
Over the years we have sequenced negative controls, consisting solely of reagents used in the lab, and with no added sample template, alongside human microbiota studies and often found varying levels of contaminating environmental bacteria in the sequence results. These include soil and water inhabitants such as Ralstonia, Bradyrhizobium, Herbaspirillum, Pseudomonas, and Burkholderia. We believed that they originated in the DNA extraction kits or the PCR reagents, and older publications confirm that this has been observed elsewhere too [1, 2].
Recently we have also seen a number of publications describing bacteria from low-biomass environments, where similar bacteria to those we had previously observed as contaminants in negative controls were highlighted as important (for example, causing a disease).
Often it was unclear whether negative sequencing controls were included in these studies or whether any contamination-checking had been done. Sometimes these publications are refuted, such as in the case of a novel virus linked to seronegative hepatitis, which actually came from a lab kit component [3].
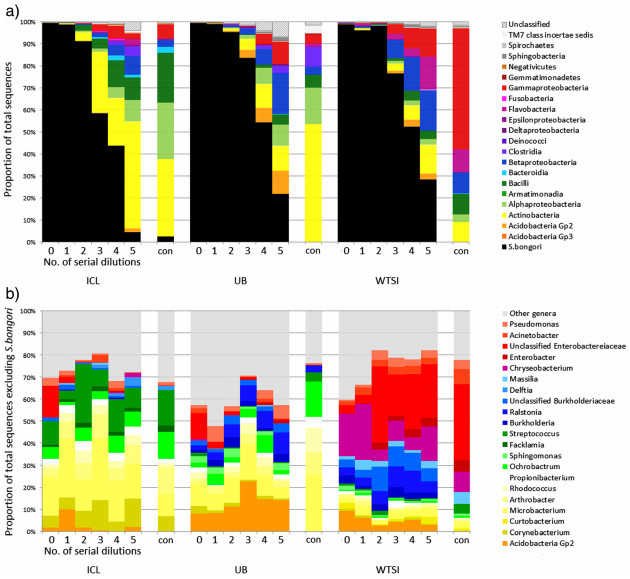
We therefore decided to sequence a “pure” sample by culturing Salmonella bongori and making a dilution series. If there was no contamination, then it wouldn’t matter how dilute the sample became, we would only ever observe one species. However, if there was contamination, we wanted to see whether there was a tipping point where the background levels of contaminant DNA became dominant over the actual S. bongori content.
To rule out the possibility that contamination issues were confined to a single DNA extraction kit manufacturer, we extracted DNA from the dilution series with four different manufacturers’ kits and compared them.
Low biomass microbiome samples are particularly susceptible
In our results, rather than just detect S. bongori, we saw a huge variety of contaminant bacteria, many of them soil/water derived but also some skin colonisers. The more diluted the samples were (i.e. the lower the amount of bacterial biomass was added) the more the contamination dominated the sequencing results.
Contaminant DNA became dominant in diluted samples that contained approximately 10,000-1,000 S. bongori cells per ml or less. This is actually quite a lot of bacteria, showing that although the contamination issue is particularly acute for low-biomass samples, it can apply to many others.
We then demonstrated the impact that contamination can have on real microbiota datasets by studying results from a sequence-based survey of the bacteria in nasopharyngeal samples. We were able to show that, although we thought we had initially observed a biologically interesting pattern, the pattern was actually caused by differing contaminant bacteria between batches of DNA extraction kits.
Conclusions
The most important conclusion, we believe, is to be prepared for DNA contamination. It is far preferable to anticipate it early on in the experiment than to discover biologically unexpected bacteria in the data only at the end.
By collecting and sequencing copious control samples (storage media controls, extraction kit controls, PCR controls, etc) if any contamination occurs during the sample handling process, there will be evidence of it in the sequence data, and the offending taxa may be screened out (with appropriate caution). Alternatively, it may be possible to remove DNA from some reagents using UV treatment, enzymes or other means.
With a greater understanding of the problem of DNA contamination in deep sequencing microbiota studies we hope it will also improve the robustness of research, as unexpected species that are implicated in disease, for example, will need to be shown to definitely exist in the samples rather than relying on genetic traces in isolation.
[1] Tanner MA, Goebel BM, Dojka MA, Pace NR: Specific ribosomal DNA sequences from diverse environmental settings correlate with experimental contaminants. Appl Environ Microbiol 1998, 64:3110-3113
[2] Grahn N, Olofsson M, Ellnebo-Svedlund K, Monstein HJ, Jonasson J: Identification of mixed bacterial DNA contamination in broad-range PCR amplification of 16S rDNA V1 and V3 variable regions by pyrosequencing of cloned amplicons. FEMS Microbiol Lett 2003, 219:87-91
[3] Naccache SN, Greninger AL, Lee D, Coffey LL, Phan T, Rein-Weston A, Aronsohn A, Hackett Jr A, Delwart EL, Chiu CY: The perils of pathogen discovery: Origin of a novel parvovirus-like hybrid genome traced to nucleic acid extraction spin columns. J Virol 2013, 87:22
Latest posts by Guest (see all)
- Microbial forensics: It’s not just fingerprints that can be left behind - 12th May 2015
- How can ‘conservation genomics’ help the recovery of the most endangered species? - 12th December 2014
- Acetate helps hypoxic cancer cells get fat - 11th December 2014
[…] A study published today in BMC Biology has found that many published microbiome studies may have been contaminated. In this guest post, Susannah Salter and Alan Walker, authors on the paper, tell us more about what they found. The last decade has seen amazing developments in DNA sequencing technology. One area that has benefitted tremendously from these advances is the field of microbiology, as it is now possible to characterise microbial communities (“microbiota”) at previously unimaginable […]
Accuracy and purity is essential, I agree with this thesis.