The birth prevalence of mitochondrial disease is approximately 1 in 5000, however 1 in 500 carry mutations in their mitochondrial genome. As there is no cure for mitochondrial disease, the immense variability in genetic defects and disease severity has made effective disease management challenging. Therefore prevention of genetic disease transmission from mother to a child using mitochondrial replacement therapy holds significant promise. Mitochondrial replacement therapy involves combining the nuclear DNA from the mother with healthy mitochondria from a donor egg. This technique was originally developed in mice, validated in primates, and recently the birth of the first ‘three person’ baby was reported in Mexico.
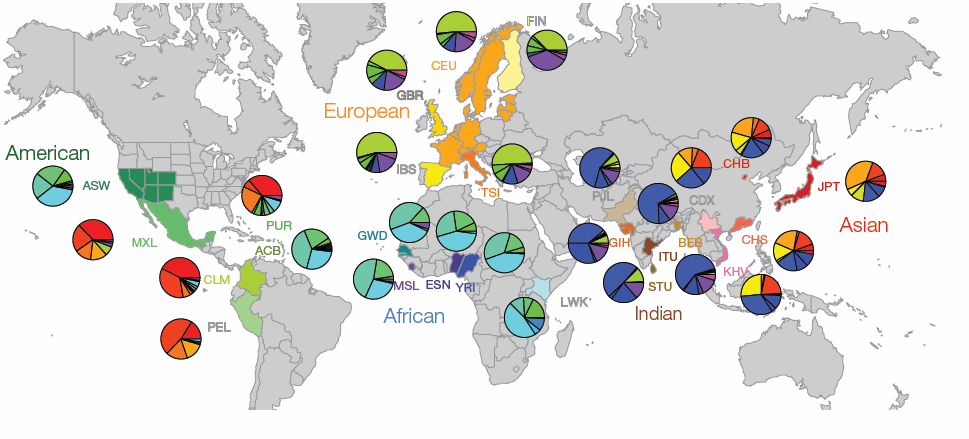
However, evolutionary biologists have raised concerns about the safety of mitochondrial replacement which are largely centered on the idea that nuclear and mitochondrial genomes evolved concurrently, and therefore mitochondria from one individual or population may not be compatible with nuclear material from another. Indeed as human populations have migrated and become distinct, as have the distribution of mitochondrial gene groups inherited from the mother (mitochondrial haplotypes). This is illustrated in the figure from the article (below), which shows the global distribution of mitochondrial DNA (mtDNA) haplogroups and highlights the immense variability and how different populations are dominated by distinct regional haplotypes.
Deleterious implications of incompatibility and nuclear-mitochondrial mismatch have been indicated by studies in model organism such as mice and invertebrates. For example, although mice with mismatched mitochondrial and nuclear genomes survived into adulthood they showed reduced physical performance, learning and growth. However, as these animals are generated by repeated inbreeding, divergent mitochondria are introduced into highly inbreed lines, and therefore these model organism studies do not reflect the human condition where breeding between populations is common.
The study recently published in BMC Genomics therefore aimed to gain an idea of the extent of nuclear-mitochondrial divergence that can be tolerated in a single individual and gain an idea of how often ‘swapping’ has occurred in human evolution.
Using data from the 1000 Genomes Project and Human Genome Diversity Project, the researchers were able to observe and quantify the extent of naturally occurring nuclear-mitochondrial mismatch seen in 3,547 healthy individuals across over 58 populations on 5 continents. As human genomic diversity has been shaped by natural selection, harmful combinations of nuclear-mitochondrial genomes should have been eliminated and therefore not observed in the existing healthy population sampled in this study.
Analysis showed that generally the genetic distance between nuclear DNA (nDNA) and mtDNA sequences, which is reflective of evolutionary relationships and migration, were highly correlated. This is what would be expected based on their co-evolution in different human populations. However, analysis also showed that even individuals with very similar nDNA genomes can have highly divergent mtDNA. For example, in Puerto Rico 67% of the sampled population have mtDNA of Native American origin and 13% carry mtDNA haplotypes of European origin. In contrast, analysis of nDNA in the same population showed the reverse with 72% European ancestry and only 13% Native American ancestry.
These findings demonstrated that mitochondrial and nuclear genomes from divergent populations can coexist in healthy individuals, indicating that mismatched nDNA/mtDNA combinations are not deleterious or subjected to purifying selection. This holds great significance for the future prospects of this technique as it suggests nuclear-mitochondrial mismatches are not likely to jeopardize the or safety of mitochondrial replacement therapy.
Comments